Timothy J. Doherty, Michael W. Nicolle
Neuromuscular Disorders*
Aging is associated with substantial decline in neuromuscular performance.1 This is perhaps best exemplified by age-associated loss of muscle mass and strength, a phenomenon often referred to as sarcopenia (see Chapter 72 for full review of sarcopenia). Neuromuscular disorders are an important cause of disability at all ages but often result in greater impairment and disability in older adults as they are superimposed on age-related impairment of both motor and sensory function of the peripheral nervous system. For example, it is well established from both anatomic and in vivo electrophysiologic studies that aging alone is associated with significant reductions in the numbers of functioning motor neurons and motor axons.1–4 This appears true for both distal and proximal muscles in the upper and lower limbs, but it may be more severe in distal lower limb muscles. Moreover, these losses of motor neurons (motor units) approach 70% in distal lower limb muscles by 80 years of age and are a major contributing factor to age-related loss of muscle mass, strength, and power (i.e., assessment of dynamic strength or force at a given velocity).5,6 In healthy older adults, lower limb strength and power are strongly related to functional indices such as gait speed and balance; these factors become of even greater importance in frail older adults.7–9 What is less well appreciated is the substantial impact on function that occurs when a disorder affecting the motor or sensory system is superimposed on the normal aging process. Often this combination results in significant disability and contributes substantially to the frailty syndrome.10 For example, an 80-year-old woman with preexisting lower limb weakness following a fall and hip fracture who develops a peroneal nerve palsy superimposed on a diabetic neuropathy will experience much greater disability as a result of these combined problems than a younger counterpart with a simple foot drop from peroneal nerve injury. In cases such as this, in addition to reduced ability to perform activities such as heavier housework or gardening, she would also walk more slowly, may have lost weight, and is likely to have reduced grip strength; muscular weakness might also contribute to a sense of excessive fatigue and increased fall risk. This is one example where a single new problem (foot drop), combined with normal age-related changes, could account for manifestation of the frailty phenotype.
The impact of aging on the sensory system is less well established. Postmortem and biopsy studies show that aging results in losses of dorsal root ganglion cells and a decline in the numbers of sensory axons.11,12 This is apparent in reduced sensory nerve action potential amplitudes from standard nerve conduction studies in older men and women.13 This likely translates to impaired sensory function that can impact balance and motor control. As with the motor system, any superimposed disorder will have a greater functional impact.10
In addition, given these observations, in some cases, slowly progressive disorders (e.g., inclusion body myositis [IBM], polyneuropathy, or polyradiculopathy) are mistaken for the expected or typical losses of muscle mass, strength, and power associated with aging. Therefore, it is imperative that clinicians appreciate common presenting features of neuromuscular disorders and recognize how they differ from so-called normal or typical aging.
To this end, this chapter focuses on disorders that are commonly found in older adults, including polyneuropathies, spinal stenosis and neurogenic claudication, myopathies, motor neuron disease (MND), and neuromuscular junction disorders. The general clinical approach is outlined, followed by a discussion of individual disorders and their treatment management.
Approach to the Patient with Neuromuscular Disease
History
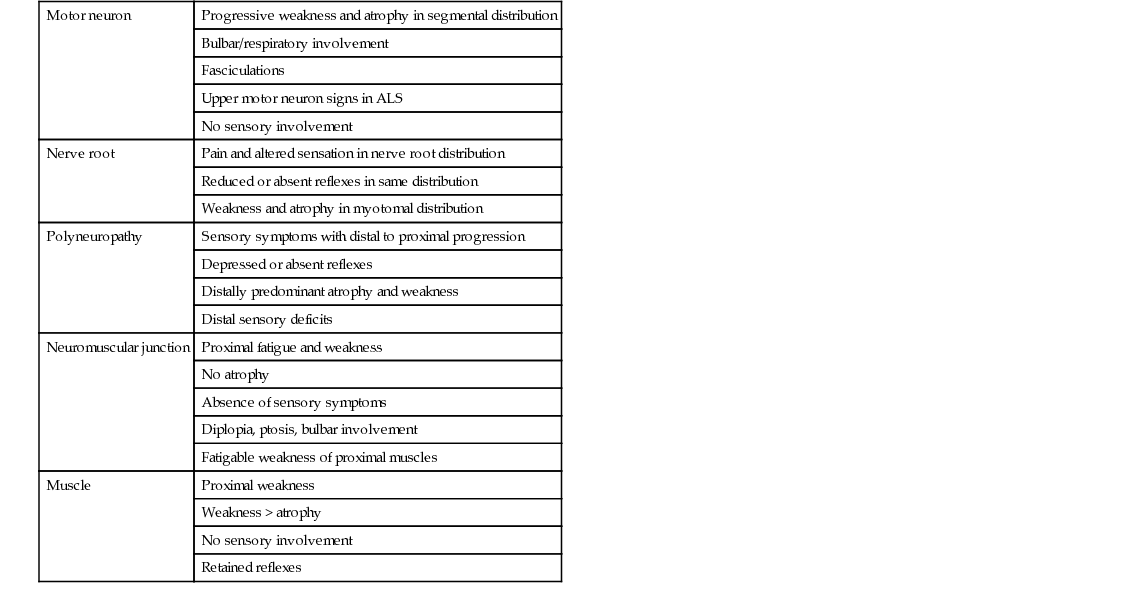
Weakness, fatigue, atrophy, and altered sensation are the most common presenting symptoms of neuromuscular disease (Table 65-1). An accurate history documenting the onset, pattern, and progression of weakness and sensory loss is crucial in differentiating diagnostic possibilities and may require several meetings with the patient and, in some cases, additional information from a spouse or relatives. In general, most myopathies and disorders of neuromuscular transmission present with proximal weakness and no sensory symptoms. Notable exceptions are myotonic dystrophy type 1 (DM1; also called Steinert disease) and IBM, which may present with predominantly distal weakness. Muscle wasting and loss of reflexes are late manifestations of most myopathies. Alternatively, most neuropathies present primarily with sensory symptoms, earlier loss of reflexes, and distal weakness. Early in the course of polyneuropathies, distal muscle wasting of intrinsic hand and foot muscles is often more impressive than strength loss. Notable exceptions are acute or chronic inflammatory demyelinating polyradiculopathy and diabetic amyotrophy, which may present with mainly proximal weakness (the latter usually accompanied by severe pain at the onset).
Inquiries into the impact of symptoms on sporting abilities, hobbies, occupational history, and military service often help establish the onset and pace of symptoms. Many patients initially ascribe their neuromuscular symptoms to normal aging or painful conditions such as arthritis, and directed questioning is often required. Questions include “How far could you walk 5 years ago?” or “When did you first use a cane or walker?” or “When could you last climb stairs?” More active patients are asked, “When could you last run?” This question is useful because increasing weakness may be present for months, and only the loss or impairment of some well-established task brings it to the patient’s attention.
The distribution of weakness is often suggested by the history: difficulties reaching up to a shelf or combing hair suggest upper limb proximal weakness. Proximal lower limb weakness is suggested by difficulty in rising from a low chair or toilet, climbing stairs, and getting in or out of the bathtub. Primary neuromuscular disease rarely presents with falls early in the course, with the exception of IBM, an inflammatory myopathy often associated with asymmetric quadriceps wasting and weakness that may include “buckling” around the knees and falls. Catching the foot on stairs or difficulty in depressing car pedals, turning a key, or opening a jar suggest distal weakness. In myasthenia gravis (MG), power may be reported as normal at rest, with fatigable weakness developing after exercise or later in the day. Fluctuation over weeks to months is also suggestive of MG and differentiates it from progressive disorders that may mimic MG, such as MND or mitochondrial myopathies. Speech and swallowing problems (including coughing and choking after ingestion of solids or liquids) and unexplained recurrent pneumonia may suggest bulbar weakness. Weakness of the cervical muscles may lead to head drop, and some patients report the need to use their hand to support the head. Some patients with cervical muscle weakness have neck pain, reflecting prolonged and ineffectual voluntary attempts to keep the head up. The causes of dyspnea are numerous and most often in older adults not primarily related to neuromuscular disorders. However, many neuromuscular disorders involve respiratory musculature. This may manifest as shortness of breath on exertion and especially on lying flat, because of diaphragmatic involvement. Inflammatory myopathies, MND, and neuromuscular transmission disorders should be considered in this setting. Other symptoms suggestive of neuromuscular hypoventilation include disrupted nocturnal sleep, daytime hypersomnolence, early morning mental clouding, and headache as a result of CO2 retention with associated cerebral vasodilation.
Myalgia is a relatively nonspecific feature seen in some patients with progressive muscular disease. Patients often find myalgia hard to describe and differentiate from joint pain. Prominent myalgia is a feature of many inflammatory myopathies, polymyalgia rheumatica, and the metabolic myopathies. However, pain at rest and lack of pain or cramp with exertion is less suggestive of an underlying defect in muscle metabolism and more suggestive of inflammatory muscle disease, referred pain from joint disease, or a myofascial pain syndrome (fibromyalgia). Uncommonly, myalgia is a presenting feature of the muscular dystrophies, such as fascioscapulohumeral dystrophy. Myotonic dystrophy type 2 (DM2; also known as proximal myotonic myopathy [PROMM]) shares some similarities with DM1 and often presents with muscle pain, stiffness, and proximal weakness. Painful nocturnal muscle cramps can reflect neurogenic diseases, including motor neuron disease/amyotrophic lateral sclerosis (MND/ALS), polyneuropathies, or chronic lumbosacral nerve root injury. Alcohol and drugs, especially those that induce hypokalemia (e.g., diuretics) and those with a structural effect on muscle (e.g., the statins), can induce myalgia. Finally, myalgia may be a prominent symptom in patients with endocrine dysfunction (especially hypothyroidism and hypocalcemia) and those with connective tissue disorders such as systemic sclerosis.
A wide-ranging systemic inquiry is essential in patients with suspected myopathies, as myositis may be a component of many collagen vascular diseases. Both DM1 and DM2 are multisystem disorders whose manifestations are varied and include diabetes, cataracts, cardiac conduction defects, and muscular weakness and wasting. Cardiac involvement is common in many neuromuscular diseases manifesting with cardiac conduction defects or cardiomyopathy or both. Prominent weight loss is a common feature in MND/ALS, reflecting both poor nutritional state and loss of muscle mass.
Many neuromuscular diseases are inherited, and therefore it is important to inquire specifically about family members and, where appropriate, about consanguinity. Premature cardiac and respiratory deaths in family members may reflect complications of an inherited neuromuscular disease or possible associated malignant hyperthermia, if associated with anesthetic exposure. It is often useful to examine first-degree relatives in a family suspected of having an inherited neuromuscular disorder even when the history does not suggest that the older relative is affected, as this can be confirmed by direct examination and has clear genetic implications for the wider family. Myotonic dystrophy, because of its marked variability in expression and the presence of anticipation, may have only minor manifestations (e.g., cataracts and mild weakness) in older adults, compared with major symptoms in siblings. The genetic defect, a trinucleotide repeat expansion, is unstable and can worsen in successive generations, particularly via the female line, leading to a phenomenon known as “anticipation,” meaning earlier onset and more severe disease in successive generations.
Sensory symptoms suggest involvement of the dorsal root ganglion, dorsal nerve roots, or sensory fibers (including the central projections such as the dorsal columns). Numbness and paresthesias distally in the toes and feet are the most common presenting symptoms of symmetric polyneuropathies. Symptoms of burning pain, coldness, tightness, and prickling may suggest predominantly small fiber involvement, whereas numbness and loss of balance may indicate predominantly large fiber involvement. The presence of orthostatic hypotension, gastrointestinal disturbance, urinary dysfunction, dryness of the eyes and mouth, and erectile dysfunction in men indicate autonomic involvement. Patchy or asymmetric sensory loss may indicate an underlying vasculitic process or sensory neuronitis. Loss of balance, particularly when in the dark (reducing visual input), may indicate large fiber sensory loss and poor proprioception. Other early clues are difficulty with balance when showering or when walking on uneven surfaces. Again, as with some early motor symptoms, these complaints are often attributed to normal aging and it is not until they are particularly disabling that medical attention is sought or investigation pursued.
Examination
The aim of the examination of the neuromuscular system is to determine the distribution of muscle weakness, sensory loss, and reflex abnormality in order to localize the lesion within the peripheral nervous system (see Table 65-1). Furthermore, it is important to assess the respiratory, cardiovascular, and dermatologic systems for associated abnormalities. The examination may provide clues to the cause and allows for grading of severity. Most acquired and inherited myopathic disorders present symptoms of proximal weakness and wasting (a limb girdle distribution). Selective patterns of muscle involvement may suggest facioscapulohumeral dystrophy (FSHD) or one of the many subtypes of limb girdle muscular dystrophy, but confirmation often relies on DNA analysis or muscle biopsy. A scapuloperoneal distribution of weakness may reflect a myopathic disorder, such as FSHD, or a neurogenic problem, such as spinal muscular atrophy. MG presents with fatigable proximal weakness but without wasting. Lambert-Eaton myasthenic syndrome (LEMS) presents with fatigable proximal weakness and wasting that can be hard to distinguish clinically from a myopathy, although the reduction in deep tendon reflexes and frequent presence of autonomic manifestations in LEMS provides a valuable diagnostic clue. Distal weakness, with involvement of the forearm and hand muscles in the upper limb and the anterior and posterior tibial compartment and intrinsic foot muscles in the lower limb, is commonly due to a peripheral neuropathy or MND/ALS but can also be seen in myotonic dystrophy, IBM, very rare distal forms of spinal muscular atrophy, and in distal myopathies. Weakness of neck flexion and extension (head drop) occurs in myopathic (e.g., DM1, inflammatory myopathy, FSHD), neuromuscular junction (MG), and neurogenic (e.g., MND/ALS) disorders. Paradoxical abdominal movements and indrawing of intercostal muscles on inspiration may indicate respiratory muscle and diaphragm weakness. Identification of isolated mild (grade 4/5 on MRC testing) weakness of the hip flexors is a common observation in older adults and often does not indicate a specific neuromuscular disorder. Therefore, when this is present, before embarking on further investigation, it is important to carefully examine other proximal muscle groups (e.g., hip extensors, shoulder girdle, neck flexors and extensors) looking for patterns of weakness that may indicate a more generalized process. It is also useful to then assess gait and ability to climb stairs or rise from a chair to determine if there are functional consequences.
Having established the pattern of weakness, the symmetry of involvement is often a guide to the underlying cause. Most myopathic diseases result in symmetric weakness. In addition, around a joint, all of the muscles will be involved to about the same degree. IBM is a noteworthy exception to this, as asymmetric forearm flexor or quadriceps involvement is common. In some neurogenic diseases, such as MND/ALS, asymmetry and unequal involvement around a joint are seen, as the weakness tends to follow a segmental pattern of spinal cord involvement, often starting locally and then progressing segmentally.
In primary muscle disorders, tone and reflexes are either normal or mildly reduced. Increased tone and reflexes suggest an upper motor neuron disorder, ALS (which ultimately has combined upper and lower motor neuron involvement), or cervical spondylitic myelopathy, the latter of which is very common in older adults and often goes unrecognized early in its course. Fasciculations are spontaneous, involuntary, visible discrete muscle twitches and reflect motor neuron or motor axon hyperexcitability. Fasciculations are not seen in muscle disease; rather, they reflect neurogenic disorders such as MND/ALS but can be seen in a wide variety of neuropathies, including focal peripheral neuropathies and chronic nerve root disease, in which denervation is a feature. As it is sometimes difficult to differentiate myopathic and neurogenic weakness on symptoms alone, a careful search should be made for fasciculations in all patients who have neuromuscular weakness. Fasciculations may be missed if patients are not undressed fully. The back, abdomen, and tongue should be inspected as well as the limbs. Difficulty is often encountered in observing fasciculations in the tongue; these are best seen with the tongue lying at rest in the floor of the mouth. Pseudofasciculations as a result of anxiety or tremor may be seen in the normal individual when the tongue is protruded.
Myotonia (delayed relaxation) is an uncommon presenting complaint in the older patient and usually signifies myotonic dystrophy, which usually presents in the second or third decade. Joint contractures are occasionally due to inherited muscle disease, whereas foot deformities such as pes cavus reflect long-standing, often genetic, peripheral neuropathies or a slowly progressive upper motor neuron disorder such as hereditary spastic paraparesis.
Depressed or absent reflexes generally indicate a neuropathic disorder, with reflex loss a late sign in muscle diseases. An exception is LEMS, in which the weakness is usually more proximal, especially involving the legs, and reflexes are reduced or absent. The combination of muscle wasting, weakness, and hyperreflexia is typical of ALS but can also be seen with cervical polyradiculopathy and concomitant cervical myelopathy. In these cases, the presence of upper motor neuron signs (e.g., jaw jerk) rostral to the most caudal lower motor neuron signs is a useful observation suggestive of ALS.
Distal, symmetric loss of sensation to pain (pinprick) and temperature are common features of typical, length-dependent neuropathies with small fiber involvement. In older adults, mild loss of vibration sense in the toes is nonspecific, but loss at the ankle or more proximally indicates large fiber involvement, as a result of either peripheral neuropathy or myelopathy with involvement of the dorsal columns. Ankle reflexes that are present or increased in the setting of significant loss of vibration in the legs are a clue to the presence of a myelopathy. Loss of proprioception is often a late finding of large fiber sensory loss, as is a positive result from a Romberg test. Obviously it is important to recognize sensory deficits that follow the distribution of individual peripheral nerves (e.g., median, ulnar, or peroneal nerve) or dermatomes because they indicate a focal mononeuropathy or radiculopathy. The combination of intermittent sensory symptoms in the hands and distal sensory loss in the feet may indicate polyneuropathy, but in older adults carpal tunnel syndrome in combination with multilevel lumbosacral nerve root compression and spinal stenosis should also be considered. As outlined later, electrophysiologic testing is invaluable in sorting these cases out.
Investigations
It is sometimes impossible on the basis of history and examination alone to make an accurate diagnosis in many cases of neuromuscular disease, not least because of the overlap in clinical signs between some neurogenic and myopathic disorders. Confirmation of a neuromuscular diagnosis requires the application of electrophysiologic, pathologic, biochemical, and, increasingly, genetic testing.
With several caveats, measurement of “muscle enzymes” is useful in patients with neuromuscular disease. Serum creatine kinase (CK) appears to be the most sensitive index of muscle necrosis from primary muscle disease and from secondary muscle fiber necrosis because of chronic denervation from neuropathic conditions. The magnitude of CK rise gives some indication of the nature of the pathology: in denervating conditions such as MND/ALS, CK levels are commonly mildly elevated in the 200 to 500 IU/L range and rarely above 1000 IU/L, whereas a more significant increase of 10- to 1000-fold suggests a primary muscle (especially inflammatory myopathy). However, CK levels must be interpreted with caution, as “muscle enzymes” are also found in other tissues. CK consists of three separate isoenzymes: MM, derived from skeletal muscle; MB, derived largely from cardiac muscle; and BB, derived mainly from brain. High CK levels may therefore be seen in patients with acute myocardial injury, large strokes, and, occasionally, hepatic disease, as well as in patients with muscle disease. Even so, given that the major isoenzyme of CK is MM, a high CK level is most likely to reflect neuromuscular disease. Finally, it is also important to appreciate that mild increases in what are typically thought to be liver enzymes, such as aspartate transaminase (AST) and alanine transaminase (ALT), can occur in primary muscle disease (ALT proportionally higher is more indicative of hepatic disease).
Electrophysiology
Electrophysiologic studies are invaluable in the diagnosis of neuromuscular disorders. A detailed discussion of electrophysiologic (electromyographic) techniques in the diagnosis of neuromuscular disease is outside the scope of this chapter but can be found in appropriate textbooks.14 Nerve conduction studies, in which the conduction velocities and amplitudes of motor and sensory (compound) action potentials in response to electric stimulation of nerves are measured, are used to detect primary pathology of peripheral nerves. Nerve conduction studies are extremely useful in detecting focal nerve injuries such as median neuropathy at the wrist in carpal tunnel syndrome, ulnar neuropathy at the elbow, or common peroneal nerve injury around the fibular head. Reduced amplitudes of motor and sensory studies with normal or mild conduction velocity slowing in the legs are typical of many of the common axonal, length-dependent polyneuropathies (e.g., drug-related, diabetes, idiopathic). Severe conduction slowing and conduction block in multiple nerve segments are important observations as they indicate an acquired demyelinating, often treatable chronic inflammatory demyelinating polyneuropathy.
The most common method for the electrophysiologic assessment of muscle is with concentric or monopolar needle electromyography (NEMG), which detects characteristic patterns that can be used to distinguish neurogenic and myopathic disorders. Normal muscle is electrically silent at rest. In neurogenic disorders resulting in denervation (e.g., ALS), spontaneous activity manifested as positive sharp waves or fibrillation potentials are seen in NEMG studies, and on voluntary activation a reduced interference pattern of the motor unit potentials is seen, reflecting the loss of motor units. By contrast, in many myopathies NEMG reveals small, short-duration motor unit potentials. Electromyography (EMG) studies may also reveal complex repetitive and myotonic (audible as a “dive bomber” or “revving motorcycle” sound) discharges, useful in confirming myotonic disorders, and may suggest a previously unsuspected diagnosis such as DM2 in which weakness predominates and myotonia is often subclinical. Spontaneous activity in the distribution of a nerve root is indicative of a radiculopathy, whereas widespread denervation in multiple regions (e.g. bulbar, cervical, thoracic, and lumbosacral) may indicate MND.
Repetitive nerve stimulation studies are useful in neuromuscular transmission disorders. In both MG and LEMS, a decrement in the compound muscle action potential response occurs with low frequency (2- to 3-Hz) stimulation, which mirrors the clinical phenomenon of fatigable weakness. In LEMS, a characteristic incremental response occurs with high-frequency (20- to 40-Hz) stimulation or after brief maximal voluntary contraction, which mirrors the clinical phenomenon of posttetanic or postcontraction facilitation. Single-fiber EMG (SFEMG) is useful in confirming a neuromuscular junction disorder, particularly in regional forms of MG. It is important to note that whereas SFEMG is highly sensitive for neuromuscular junction disorders (>95%), it is, in turn, very nonspecific with abnormal results possible in any chronic neurogenic condition or myopathy.
Muscle Biopsy
Despite advances in biochemistry, neurophysiology, and genetics, the final diagnosis in patients with muscle disease often requires a muscle biopsy. The development of the technique of needle muscle biopsy, which can be undertaken as a simple outpatient procedure, has made possible one-stop diagnostic neuromuscular clinics with combined clinical, neurophysiologic, and muscle sampling.
The vastus lateralis and deltoid are most commonly biopsied, ideally sampling a muscle that is weak but only moderately affected clinically but not too atrophied, for fear of sampling muscle with only end-stage pathology. Routine histologic stains can be employed on both paraffin-embedded and fresh frozen material and permit assessment of muscle fiber size and morphology and the presence or absence of inflammation. Other stains allow differentiation of muscle fiber types and can be used to study the distribution of cellular enzymes and metabolic reserves.15 Immunohistochemistry on frozen muscle using antibodies directed against sarcolemmal muscle proteins, such as dystrophin and the sarcoglycans, is crucial in the diagnostic workup of suspected dystrophinopathies and limb girdle muscular dystrophies and permits a more focused search for genetic abnormalities. Western blotting techniques on muscle are often essential in confirming the suspicion of muscular dystrophies. Direct measurement of enzyme activity in fresh muscle is sometimes useful in diagnosing rare metabolic disease such as acid maltase disease and in mitochondrial myopathies where respiratory chain enzymes can be assayed. Electron microscopy of muscle is useful to confirm suspected mitochondrial abnormalities seen on light microscopy and especially to look for intracellular inclusions, which occur in some inherited and acquired muscle disease.
Muscle samples are hard to process, hard to orient, and fast to degrade; for these reasons, the technique of muscle biopsy is unsuitable for routine laboratories. Furthermore, interpretation of muscle biopsies and exclusion of artifactual change is difficult. Therefore, it is important to send muscle samples to special neuromuscular laboratories or to an experienced pathology center. As percutaneous needle and punch biopsies are far less invasive than open procedures, it is possible to use sequential biopsies to follow patients with a muscle disease and monitor response to treatment in individual patients. The smaller sample sizes inherent with these techniques mean that patchy inflammation may be missed.
Peripheral Neuropathies
Peripheral neuropathies are overall the most common neuromuscular disorders found in older adults. It is beyond the scope of this text to provide an in-depth review of all peripheral neuropathies, and the reader is directed to a number of excellent texts.14,16 This section provides an overview of peripheral neuropathies and specifically addresses diabetic neuropathy because of its high prevalence in older adults.
Typical symptoms of peripheral neuropathy include distally predominant weakness, sensory loss, poor balance, pain, and autonomic dysfunction. Weakness in the majority of polyneuropathies follows a length-dependent pattern and is therefore often more severe in the lower than upper limbs. Weakness tends to become symptomatic in the extensors of the toes and ankles and evertors of the ankle earlier than in the plantar flexors. In the upper limb, difficulties with fine motor tasks such as fastening buttons or picking up coins can be early indications of weakness.
Sensory symptoms of polyneuropathies can be divided into those that indicate involvement of small, thinly myelinated fibers that subserve pain and temperature and those that suggest involvement of large myelinated fibers that are involved in position sense. Common symptoms of small fiber neuropathies include hypersensitivity to footwear or bedclothes, shooting or stabbing pain, difficulty detecting temperature of bath water, and burning sensation. These symptoms usually predominate in the feet, as most neuropathies are length-dependent. Thus, when sensory symptoms reach the level of the knees, they often begin in the hands. Small fiber sensory symptoms of burning, prickling, and allodynia are a common cause of sleep disturbance in older adults. These symptoms should prompt a careful assessment for decreased sensation to pinprick and temperature for consideration of a small fiber–predominant neuropathy.
Large fiber involvement, particularly if it is severe, will typically present with loss of balance and gait difficulty because of the loss of proprioception or position sense. These symptoms often result in mobility limitations10 in older adults and fear of falling. In particular, patients with balance impairment secondary to peripheral neuropathy tend to avoid crowded areas, such as grocery stores and shopping malls. These symptoms should prompt an examination for decreased sensation to light touch, vibration, and position sense as well as reduction or loss of deep tendon reflexes.
Autonomic symptoms include urinary retention or incontinence, abnormal sweating, constipation and diarrhea, and symptoms of orthostatic hypotension.17 These symptoms are often initially overlooked as indicative of a neuropathic disorder and prompt assessment for a primary cardiac or central neurologic disorder cause.
Most neuropathies affect both motor and sensory fibers; however, pure or predominantly sensory involvement can be seen in concert with diabetes, malignancies (paraneoplastic neuropathies), and idiopathic sensory neuropathy in older adults. Pure motor involvement may indicate multifocal motor neuropathy (MMN),18 a rare demyelinating disorder that typically presents initially with focal weakness of upper limb muscles, or may suggest MND. Along the same lines, most neuropathies have symmetric, distally predominant features. Asymmetry may indicate mononeuritis multiplex associated with vasculitis, hereditary neuropathy with liability to pressure palsies, or common focal or entrapment neuropathies. Box 65-1 outlines some common presentations based on the predominant fiber population involved.
Once a pattern of small or large sensory fiber predominance has been established, the absence or presence of motor involvement has been defined, and the symptoms and signs have been determined as symmetric or asymmetric based on the clinical assessment, it is often of considerable value to obtain electrophysiologic studies before other investigations to confirm or extend the clinical characterization. Most expert clinicians agree that is also useful to obtain a fasting blood sugar level, serum creatinine level, electrolyte panel, complete blood count, vitamin B12 level, and serum protein electrophoresis (and immunofixation if indicated) as part of the initial workup.19 Often expensive testing for specific antibodies or genetic testing should await the results of electrophysiologic testing and is often best directed by clinicians and centers with specialized expertise.
Electrophysiologic testing is extremely helpful in tailoring future investigation because it can determine whether the process is predominantly axonal (most common) or demyelinating as well as reveal subclinical motor or sensory involvement. The presence of a demyelinating process is extremely important to establish, as this often indicates a treatable acquired neuropathy (e.g., chronic inflammatory demyelinating polyneuropathy or MMN) or a hereditary neuropathy if there is uniform slowing of conduction velocities. If an axonal neuropathy is present, it is important to determine if it is symmetric (most common) or asymmetric and multifocal, which may indicate an underlying vasculitic process requiring further investigation and treatment.20
It is important to note that standard nerve conduction studies examine only large, myelinated fibers. Therefore, the results of the studies may be normal or only mildly affected in small fiber neuropathies (e.g., diabetes).
Diabetic Neuropathy
Diabetic neuropathy is the most common form of peripheral neuropathy in the Western Hemisphere, with increasing prevalence resulting from the growing prevalence of obesity and type 2 diabetes.21 A number of different classification schemes exist for diabetic neuropathy; a common one is outlined in Box 65-2. The most common form is a mixed but predominantly sensory, motor, and autonomic symmetric diabetic peripheral neuropathy (DPN), which may comprise up to 70% of cases.15
A predominantly sensory, often painful, neuropathy comprises the other largest group. Diabetic neuropathy is common and may be present in up to 50% of individuals with type 1 diabetes and 45% of those with type 2 diabetes if comprehensive batteries of testing are used.21,22 A general rule is that the prevalence of a neuropathy in diabetes increases 1% to 2% for each year a patient has diabetes. In patients with only impaired glucose tolerance, the prevalence figures remain controversial.23 Risk factors for DPN include the duration and severity of hyperglycemia, smoking, other complications such as retinopathy or nephropathy, and cardiovascular disease. The pathophysiology of DPN remains somewhat controversial but includes axonal injury from hyperglycemia and associated polyol flux, particularly sorbitol through the aldose reductase pathway; microangiopathy and hypoxia; oxidative and nitrative stress from free radicals; and deficiency of growth factors.15 Indeed, the metabolic syndrome itself may be directly linked to diabetic neuropathy through the combined effects of dyslipidemia, insulin resistance, systemic inflammation, and the activation of the renin-angiotensin-aldosterone system leading to oxidative stress and cellular damage.21
Symptomatically, patients with DPN typically have positive neuropathic features such as prickling, tingling, and pins and needles; burning; or, occasionally, shooting sensations. Negative symptoms such as numbness of the toes or feet can paradoxically occur along with the positive features. Many patients experience symptoms mainly at night and experience painful allodynia (pain in response to nonpainful stimuli) from bed sheets; others experience symptoms throughout the day that are related to walking or footwear. True sensory ataxia is less common but can occur with severe involvement. Symptoms may stay confined to the lower extremity but may advance to the hands as they progress to the level of the knees in the lower limb. Early sensory symptoms in the hands should raise the question of a superimposed carpal tunnel syndrome, which has a very high prevalence in those with DPN.24
Clinical examination in patients with DPN reveals distal, sensory greater than motor deficits to all sensory modalities and often loss of ankle deep tendon reflexes. Motor deficits are less common, but patients may have weakness of toe extensors and flexors and, in more severe cases, weakness of ankle dorsiflexors.
The most effective intervention to prevent the incidence or limit the progression of DPN is enhanced glucose control. The Diabetes Control and Complications Trial followed more than 1400 individuals with type 1 diabetes for 5 years and reported a 60% reduction in the incidence of DPN in those receiving more frequent insulin dosing.25 Similarly, Linn and colleagues reported a 70% reduction in DPN in 49 patients treated with enhanced glucose control for 5 years.26 In contrast, the benefits of enhanced glucose control have been less definitive in those with type 2 diabetes.21
Diabetic foot ulcers are of considerable importance when assessing older adult patients with DPN. Diabetic ulcers occur because of a combination of sensory loss and repetitive pressure on bony prominences such as the metatarsal heads or heel. This, in combination with trophic changes caused by the neuropathy, leading to drying and cracking of the skin, leads to chronic tissue injury. Further progression may occur as a result of loss of proprioception leading to abnormal foot position and biomechanics. Careful inspection of the feet on a daily basis, screening for early evidence of sensory deficits, proper footwear with adequate height of the toe box or forefoot, and foot orthoses are all useful in terms of preventing the occurrence of ulceration and reducing the risk of amputation.27
Treatment of neuropathic pain associated with DPN has been the topic of numerous well-controlled clinical trials. Well-established guidelines support the use of a number of pharmacologic approaches with the strongest evidence in favor of tricyclic antidepressants, serotonin-norepinephrine reuptake inhibitors, pregabalin, and gabapentin.28
Diabetic lumbosacral radiculoplexopathy neuropathy (DLRPN), often referred to as diabetic amyotrophy or proximal diabetic neuropathy, requires special mention because of its higher prevalence in older adults and the severe disability often associated with it. DLRPN is a devastating condition that affects only about 1% of individuals with diabetes, more commonly type 2.29 It typically presents with severe, asymmetric, acute onset of proximal leg pain and weakness. Frequently it occurs in concert with a large concomitant weight loss. In many cases, affected patients have not been diabetic for a long period of time and have no other end organ complications from DM, including frequent absence of a length-dependent diabetic neuropathy. The symptoms are usually unilateral or asymmetric and involve proximal lower limb segments such as the hip flexors and knee extensors. The condition may spread more distally and to the contralateral side over a few days. Although pain is the most severe initial manifestation, often requiring narcotic analgesia, severe weakness typically develops in the first few days, often severely affecting the hip flexors and knee extensors and also the more distal muscles, including the ankle plantar and dorsiflexors. Gait aids or wheelchairs are often required for mobility.
The cerebrospinal fluid may reveal elevated protein, providing evidence that the disease process is proximal at the level of the spinal roots. Electrophysiologic (EMG) testing reveals axonal injury or denervation in affected muscles, often including the paraspinal muscles, often with severe loss of recruitment implying substantive loss of axons. Given that axonal loss is the mechanism, the time course of recovery is typically many months. In our experience, most of these patients do well and improve gradually, if provided supportive therapy and then treated later with appropriate physical therapy in the form of resistance exercise and gait retraining.
The purported pathophysiologic basis of DLRPN is ischemic injury, possibly secondary to microvasculitis. Given this, immunomodulation may be useful if started early in the course of disease, as has been demonstrated in nondiabetics with idiopathic lumbosacral plexopathy. To date, however, clinical trials in people with diabetes have not supported this theory.29
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


