Clinical scenario
Recommendations
Level of evidenceb
Spinal cord compression
Corticosteroids: decadron 1–2 mg/kg (max 10 mg) followed by 0.25–0.5 mg/kg (max 4 mg) Q6h—may complicate ability to make a diagnosis
1C
Surgery: rapid progression of symptoms or total plegia are indications for emergent surgical management
1B
Radiation therapy: radiosensitive tumors (i.e., lymphoma, leukemia) while leaving additional diagnostic sites; patients who are too unstable for surgery; for palliation
1C
Altered mental status
Workup to include structural, metabolic and infectious causes
1C
Antiepileptics (benzodiazepines first-line) for acute seizure management with AMS
1B
Hypertonic saline for seizures secondary to hyponatremia, then slow correction to avoid central pontine myelinolysis
1B
Increased intracranial pressure
Acute evaluation and management of airway, breathing and circulation in the intensive care setting
1C
Consideration for intubation
1C
Elevate head of bed
1C
Treatment of hyperthermia with antipyretics and cooling
1C
Head CT or MRI prior to LP
1C
Measurement of LP opening pressure
1B
Consideration for hyperosmolar therapy with mannitol or hypertonic saline
1C
Cerebrovascular accident
MRI preferred over CT; CT can be done acutely to rule out hemorrhage
1C
Treatment with a specialized team including pediatric stroke specialist, neurosurgeon, interventional radiologist
1C
Permissive hypertension in ischemic stroke
1C
tPA if no contraindications in ischemic stroke
2C
Assessment for underlying prothrombotic state
2C
Neurosurgical intervention for hemorrhagic stroke
1C
Assessment of coagulopathy and FFP therapy if coagulopathic
1C
Utilization of recombinant factor VII in hemorrhagic stroke
2C
Sinovenous thrombosis secondary to asparaginase therapy
Measurement and repletion of antithrombin (with pooled or recombinant product) and plasminogen
1B
Repletion of fibrinogen with cryoprecipitate to normal levels (i.e., >150 mg/dL)
1C
Anticoagulation if without a severe hemorrhagic component (i.e., unfractionated heparin)
1C
Reinstitution of asparaginase after recovery
1B
Prophylaxis with antithrombin is not currently recommended
2C
Neurosurgery
Prophylactic antiepileptics after neurosurgical intervention
2C
IT neurotoxicity
Dextromethorphan in the acute phase of IT MTX neurotoxicity
2C
Aminophylline in the acute phase of IT MTX neurotoxicity
2C
Leucovorin rescue for patients who have had IT MTX neurotoxicity in the past
2C
Rechallenging with IT MTX or substituting with IT HC/Ara-C for future IT therapy
2C
Ifosfamide-induced encephalopathy
Methylene blue for acute treatment or future prophylaxis if with IIE
2C
Thiamine for acute treatment or future prophylaxis if with IIE
2C
Posterior reversible encephalopathy syndrome
Acute seizure management (if present) as above
1B
Controlling of blood pressure with calcium channel blockers or β-blockers
1B
MRI imaging with FLAIR and DWI
1B
5.2 Spinal Cord Compression
Compression of the spinal cord can be caused by mass effect from either extradural or intradural tumors. Presenting symptoms can initially be nonspecific, and the diagnosis can be missed if the index of suspicion on the part of the examiner is not sufficiently high. Cord compression may herald a new diagnosis of cancer, be a sign of treatment failure or relapse, or indicate a complication of treatment such as a post-lumbar puncture hematoma. In addition to spinal cord compression, this section describes conus medullaris and cauda equina syndromes.
5.2.1 Presentation
Back pain is a presenting symptom in 80–90 % of patients presenting with malignant spinal cord compression (Pollono et al. 2003). In patients old enough to describe their symptoms, this pain is classically radicular, radiating to the legs, sharp, and described as electric. Non-radicular pain may be also reported. However, younger patients may not be able to localize pain or provide any description of its quality.
Weakness is present in similar numbers of patients at presentation, but again can be difficult to elicit in the young child. Increased “clumsiness,” particularly affecting gait, or refusal to walk, can be manifestations of neurologic weakness. In patients who are able to comply with examination, localization of the weakness can help identify the level of compression (Table 5.2). Weakness may initially be flaccid, with subsequent hyperreflexia in extremities distal to the site of compression. Compression at the level of the cauda equina causes absent reflexes due to the peripheral nature of the injury.
Table 5.2
Localization of spinal cord compression by weakness
Nerve roots | Muscle action and deep tendon reflexes (DTRs) |
|---|---|
C3–C5 | Diaphragmatic excursion |
C5–C6 | Biceps and brachioradialis DTRs |
C6–C7 | Triceps DTR |
C8–T1 | Hand function |
L2–L3 | Hip flexion |
L3–L4 | Patellar DTR |
L4–L5 | Ankle dorsiflexion |
L5–S1 | Knee flexion |
S1–S2 | Ankle plantarflexion; S1 mediates Achilles DTR |
Isolated bowel or bladder dysfunction with perineal anesthesia suggests compression of either the conus medullaris or cauda equina, but sympathetic denervation from a higher compression can reproduce symptoms of urinary retention. A history of progressive constipation or abdominal pain with a full bladder should therefore warrant further investigation. Sensory changes (paresthesias or anesthesia) should be carefully investigated to determine patterns of distribution. The presence of any sensory level of such findings is a very concerning sign and can additionally help localize a lesion.
5.2.2 Differential Diagnosis
Cord compression can be the presenting feature of a new malignancy and patients may have either acute or subacute presentations. Of tumors arising from the extradural space, paravertebral Ewing sarcoma is the most frequent malignancy to cause cord compression (Klein et al. 1991; Pollono et al. 2003). Neuroblastoma can also present with cord compression given their origin in the prevertebral sympathetic chain. While less common, primary or metastatic vertebral bone lesions such as osteosarcoma or Langerhans cell histiocytosis have also been associated with cord compression. Pathologic fracture complicating a primary bone lesion may cause the presenting symptoms. Both acute lymphoblastic leukemia (ALL) and acute myelogenous leukemia (AML) can present with extramedullary disease that can cause cord compression. Discrete masses composed of leukemic cells (chloromas or granulocytic sarcomas), are more commonly observed in AML than ALL (Mantadakis et al. 2008; Olcay et al. 2009; Isome et al. 2011). Both Hodgkin and non-Hodgkin lymphoma can similarly lead to cord compression through direct mass effect (Acquaviva et al. 2003; Daley et al. 2003; Gupta et al. 2009).
Intraspinal tumors in children (Fig. 5.1) are most commonly gliomas or ependymomas (Huisman 2009; Benesch et al. 2010). Drop metastases from posterior fossa tumors, most notably primitive neuroectodermal tumors (PNET; medulloblastoma), can also cause cord compression. Patients with neurocutaneous syndromes such as neurofibromatosis type-1 (NF-1) and NF-2, von Hippel-Lindau disease, and tuberous sclerosis warrant special consideration when presenting with signs of spinal cord compression given their propensity for various intraspinal tumors such as malignant peripheral nerve sheath tumor, ependymoma, hemangioblastoma and astrocytoma, respectively.
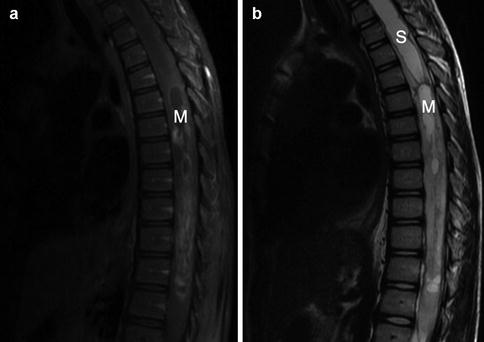
Fig. 5.1
Intraspinal glioma. This 10-year-old patient presented with several weeks of progressive bilateral lower extremity weakness. The T1-weighted gadolinium-enhanced MRI shown in Panel a demonstrates a cystic mass (“M”) in the upper thoracic spinal cord. The T2-weighted image in Panel b highlights the development of syrinx (“S”), seen as bright cerebrospinal fluid in this sequence, cranial to the mass. Pathology from a biopsy specimen taken at the time of diagnosis was consistent with pilocytic astrocytoma
In the patient with known tumor, new symptoms of cord compression should raise concern for development of metastatic disease. Patients who have recently undergone a lumbar puncture, particularly in the setting of low platelets or coagulopathy, are at risk for an epidural hematoma that may require emergent evacuation. A potential mimic of cord compression in the pediatric oncology patient is acute onset transverse myelitis. This entity can present with symptoms of weakness, bowel and bladder dysfunction, and a sensory level; magnetic resonance imaging (MRI) may demonstrate inflammatory changes within the cord, but no compressive mass. Transverse myelitis has been reported in patients following either intrathecal or high-dose intravenous cytarabine (Schwenn et al. 1991).
5.2.3 Management
The first step in managing a patient with suspected cord compression is involvement of a multidisciplinary team including neurosurgery, neurology, oncology, intensivists and radiation oncology. Frequent monitoring for progression of symptoms is vital, as this can rapidly change the therapeutic plan. Initial steps should be aimed at verifying the diagnosis and attempting to alleviate compressive symptoms medically while a more definitive treatment plan is formulated.
5.2.3.1 Imaging
MRI of the spine can help narrow the differential diagnosis, allow for surgical planning and identify potential sites of multifocal disease. The entire spine should be imaged with pre- and post-gadolinium contrast to maximize yield. Brain imaging may also be warranted if the lesions are suspicious for drop metastases or if the patient presents with signs or symptoms of increased intracranial pressure such as headache, emesis, hypertension with bradycardia, or cranial nerve palsies.
5.2.3.2 Corticosteroids
Steroid treatment is employed to relieve any vasogenic edema that may be contributing to cord compression. In the case of ALL and lymphoma, corticosteroids also have a direct antitumor effect that may benefit the patient in the short term, but ultimately make diagnosis of the underlying malignancy much more challenging. If a hematopoietic malignancy is likely, steroids should be given only when urgent diagnostic procedures have been scheduled (e.g., bone marrow aspiration and biopsy, lymph node excisional biopsy).
Outside of this scenario, the benefits of steroids are thought to outweigh their risks and are routinely used when cord compression is diagnosed, although clinical trials in children are lacking. In adults there is no evidence supporting high-dose dexamethasone; an initial dose of 1–2 mg/kg (max 10 mg) followed by 0.25–0.5 mg/kg (max 4 mg) every 6 h can be considered while awaiting further therapy (Kaal and Vecht 2004; Loblaw et al. 2005; George et al. 2008).
5.2.3.3 Surgery
Surgery in spinal cord compression can be utilized for tumor debulking with spinal cord stabilization or for diagnostic biopsy in the highly chemotherapy- or radiotherapy-sensitive tumor. Prospective studies in adult patients with cord compression have favored early laminectomy and debulking over radiation therapy alone although more recent retrospective studies have called this into question (Patchell et al. 2005; Rades et al. 2010). Laminectomy in the young child can lead to spinal deformities requiring future surgical intervention and therefore pediatric retrospective studies argue against an early surgical approach (Parikh and Crawford 2003). Recovery from surgery can also lead to delays in instituting definitive chemoradiotherapy.
An early case series from St. Jude Children’s Research Hospital suggested that initial debulking is warranted for sarcomas, whereas biopsy with chemotherapy or radiotherapy should be considered for patients with neuroblastoma, germ cell tumors and lymphomas (Klein et al. 1991). Patients presenting with rapid evolution of symptoms or complete loss of motor function were treated with urgent surgical decompression regardless of tumor histology. Smaller case series have supported the use of initial chemotherapy in neuroblastoma, Ewing sarcoma and germ cell tumors (Hayes et al. 1984). A study of 76 children with neuroblastoma from Italy found no difference in the success rates of chemotherapy, radiotherapy or surgical debulking in improvement of neurological signs, but noted that patients receiving radiotherapy or surgical debulking all went on to receive additional therapy for cord compression, whereas chemotherapy patients did not (De Bernardi et al. 2001). A follow-up study from this same group also noted a higher incidence of spinal deformities in the group receiving surgical intervention with a trend towards worse neurologic outcomes. As with the St. Jude data, patients with rapidly progressive symptoms or severe presentations were all treated with emergent surgery, making that group much higher risk than those treated with chemotherapy or radiotherapy (Angelini et al. 2011). A more contemporary study of 122 patients did not find any difference in spinal outcomes in patients treated with chemotherapy or surgery which the authors attribute to improvement in surgical technique (Simon et al. 2012).
In summary, each case of cord compression warrants individual consideration before a decision to proceed with debulking or biopsy can be made. Rapid progression of symptoms and total plegia are indications for emergent surgical management. Spinal column instability, which warrants surgical intervention, will also often necessitate surgical debulking. If the malignancy is known to be chemotherapy sensitive (i.e., hematopoietic malignancies, neuroblastoma, Ewing sarcoma, germ cell tumors), biopsy either at the site of cord compression or at another more easily accessible site can be considered prior to institution of chemotherapy. The timely availability of these options may also influence the treatment decision.
5.2.3.4 Radiation Therapy
The advantages of external beam radiation therapy include rapid onset of action and minimal invasiveness making radiation therapy an attractive option for patients who are too unstable to be considered surgical candidates, or in the palliative setting. Tumors that are radiosensitive such as hematologic malignancies (i.e., chloromas) and lymphoma may benefit from urgent radiotherapy while leaving additional diagnostic disease sites intact. Extensive metastatic disease may similarly be more amenable to radiation therapy than a surgical approach; when surgery is the primary therapeutic modality, adjunctive radiation may have a role in local control.
A case series from Children’s Hospital of Philadelphia demonstrated the benefit of urgent radiation therapy in combination with chemotherapy in a pediatric population with a variety of tumor types: of 33 patients presenting with cord compression, 55 % demonstrated improvement following chemoradiotherapy and 30 % had stabilization of their neurologic symptoms (Bertsch et al. 1998).
Dose-dependent long-term complications arising from radiation therapy can include future growth complications such as scoliosis as well as risk of secondary malignancy, particularly meningioma (Hoffman and Yock 2009). Stereotactic radiosurgery can deliver a single treatment dose up to 13 Gy to localized tumors without exceeding spinal cord tolerance; additionally, the spinal cord can tolerate high total doses, with a risk of myelopathy of <1 % at 54 Gy in adult patients (Kirkpatrick et al. 2010).
5.2.4 Outcomes
The best predictor of outcome in multiple studies has been the degree of neurologic disability at the time of presentation. One large cohort study suggested that only approximately 50 % of patients with paraplegia at the time of presentation improved despite treatment whereas close to 90 % of patients without paraplegia had improvement in their symptoms (Pollono et al. 2003). Published outcomes data are from small cohort studies with a variety of pathologic diagnoses and treatments, making any broad interpretation difficult. There is a general consensus, however, that outcomes for pediatric cord compression are significantly better than in adult patients.
5.3 Altered Mental Status
For the purposes of this chapter, altered mental status (AMS) includes patients with symptoms of encephalopathy (confusion, somnolence or coma) and those with seizures (which often, though not always, alter the level of consciousness). Although there are often distinct differential diagnoses for encephalopathy and seizure, many conditions unique to the pediatric oncology patient can predispose to either presentation. Additionally, subclinical seizure is a diagnostic consideration in any patient with altered mentation. For these reasons, we suggest a common approach to the differential diagnosis and initial management of such patients.
5.3.1 Presentation
Confusion or somnolence in an oncology patient should prompt a thorough neurologic examination and review of recent medications. Nonresponsive patients require urgent steps to secure the airway, breathing and circulation prior to additional diagnostic interventions. AMS may be due to a diffuse process affecting the brain, such as medication effect or subclinical seizure, or be due to direct involvement of the brainstem, increased intracranial pressure (ICP), or impending herniation. Ruling out the latter by means of neurologic assessment is of utmost importance. Details in the management of increased ICP are discussed in Sect. 5.4.
5.3.2 Initial Management
The first goal in a seizing patient is to stop the seizures. Benzodiazepines are often used as first-line medications in this setting. Lorazepam can be given intravenously if access is available; otherwise intranasal, buccal, or rectal formulations of diazepam or lorazepam can be employed. It is important to anticipate and treat the adverse effects of these medications including respiratory depression or hypotension, as repeated dosing may be required to halt seizure activity. Failure to respond to benzodiazepines (or recurrent seizures after initial response) should prompt escalation to additional antiepileptics such as phenobarbital or fosphenytoin. Levetiracetam is another antiepileptic that has the advantage of not inducing hepatic enzyme activity and thus has fewer interactions with chemotherapy. Seizures refractory to these interventions can be treated with intravenous loading with either valproic acid or levetiracetam and necessitate urgent determination of the underlying etiology.
Initial diagnostic workup should include serum chemistries, especially sodium, calcium, magnesium, phosphorus, and glucose, and head imaging. CT is often the easiest imaging to obtain in this setting and can rule out emergent, life-threatening causes of seizure. If there are concerns for cerebrovascular accident, a stroke-protocol MRI with diffusion-weighted imaging or magnetic resonance angiography should be considered. The seizing oncology patient should be managed in consultation with neurology; early neurosurgical involvement is also imperative for patients with known or suspected intracranial processes.
Continuous electroencephalography (EEG) monitoring should be strongly considered for any patient with persistent AMS or concerns for subclinical seizure. Lumbar puncture and MRI may be indicated when the patient is stabilized to look for evidence of progressive malignancy, infection or demyelination. Further treatment is dependent on the results of these initial diagnostic studies, as discussed below.
5.3.3 Differential Diagnosis
The differential diagnosis of seizure or AMS can be broadly divided into structural causes, including primary CNS or metastatic disease; metabolic derangements resulting in hyponatremia, hypocalcemia or hypoglycemia; infectious complications; toxic effects of chemotherapy; or stroke (ischemic or hemorrhagic). Posterior reversible encephalopathy syndrome (PRES) can also present with seizure. Seizures secondary to neurologic emergencies such as stroke are discussed in greater detail in Sect. 5.5.
5.3.3.1 Structural
Primary CNS tumors can directly lead to seizures. Oligodendrogliomas and gangliogliomas are common causes of brain tumor-induced seizure, though other low-grade gliomas and dysembryoblastic neuroepithelial tumors (DNETs) can be associated with seizures as well (Ogiwara et al. 2010). Such low-grade lesions may be surgically resected to provide relief from symptomatic seizures. Extracranial tumors can invade the CNS either by direct invasion, as with parameningeal rhabdomyosarcoma, or by hematogenous metastasis, as in Ewing sarcoma (the most common cause of pediatric CNS parenchymal metastasis), extracranial germ cell tumors, and leukemias.
Intracranial hemorrhage is another consideration in the seizing patient. Intratumoral hemorrhage has been variably reported in adults receiving the monoclonal antibody bevacizumab for primary CNS tumors (Seet et al. 2011; Khasraw et al. 2012). Certain primary childhood CNS tumors have a high proclivity for spontaneous intratumoral hemorrhage due to their intrinsic high vascularity; notorious examples are primary CNS choriocarcinoma, choroid plexus carcinoma and malignant gliomas. Derangement of the hemostatic system as seen in acute promyelocytic leukemia or after asparaginase therapy can also lead to intracranial hemorrhage in a patient without CNS disease.
Patients who have undergone resection of CNS tumors may develop postoperative seizures. No clear consensus exists regarding the use of prophylactic antiepileptic therapy in the postoperative setting. The American Academy of Neurology recommends tapering antiepileptic drugs (AEDs) within the first postoperative week, though it does not make a recommendation regarding AED initiation due to a lack of evidence even in the adult setting (Glantz et al. 2000). A retrospective study of 223 patients from Children’s Hospital of Philadelphia found that age younger than 2 years, supratentorial location of tumor and postoperative hyponatremia were the only independent predictors of postoperative seizure (Hardesty et al. 2011). Of the 229 operations reviewed, 7.4 % of patients seized and only 4.4 % received routine postoperative AEDs (Hardesty et al. 2011). Due to insufficient evidence, the use of prophylactic AEDs should be determined under consultation with the patient’s neurosurgeon and intensive care team. Given the low incidence of unprovoked postoperative seizure, workup of new seizures following tumor resection is warranted to exclude alternative etiologies.
5.3.3.2 Metabolic
The metabolic causes of AMS or seizure in pediatric oncology are similar to other pediatric patients: hyponatremia, hypoglycemia, and, less frequently, hypocalcemia secondary to hyperphosphatemia in severe tumor lysis syndrome (discussed in Chap. 3).
Iatrogenic Hyponatremia
Hyponatremia severe enough to provoke seizures warrants urgent correction though overly rapid correction can lead to central pontine myelinolysis. The risks of this complication are greater in chronic hyponatremia and in adult patients. A reasonable goal is to raise the sodium by ≤10 mEq/L in the first 24 h and then slowly normalize the serum sodium over the next 48 h. Hypertonic saline should be used in the seizing patient as it allows for more rapid correction than isotonic fluids which can be accomplished by calculation of the sodium deficit and either a subsequent fluid infusion rate or bolus therapy (Adrogue and Madias 2000; Sterns et al. 2009; Moritz and Ayus 2010).
Syndrome of Inappropriate Antidiuretic Hormone (SIADH) Release
The syndrome of inappropriate antidiuretic hormone (SIADH) secretion leads to dilutional hyponatremia due to inappropriate resorption of free water in the renal collecting tubules. SIADH has a multitude of causes including poorly understood effects of CNS or intrapulmonary lesions as well as secondary to chemotherapeutic agents including vincristine, cyclophosphamide, cisplatin and melphalan (Lim et al. 2010). The diagnosis of SIADH relies on the combined laboratory findings of low serum osmolarity (i.e., <290 mOsm/L) with inappropriately concentrated urine osmolality (i.e., >100 mOsm/kg).
Treatment of SIADH depends on: (1) removing the causative agent when possible; (2) restricting free water intake to ≤urine output (±insensible losses); and (3) correction with hypertonic saline, if indicated. Hypertonic saline is generally avoided in patients with asymptomatic SIADH and in those presenting solely with AMS but may be necessary to control seizures. Free water restriction is difficult in situations where hyperhydration is necessary to avoid chemotherapeutic toxicity as with cyclophosphamide administration. In these situations, using normal saline as opposed to hypotonic fluids and checking serum sodium levels frequently is warranted. Chronic SIADH can be managed with oral urea and fluid restriction (Huang et al. 2006). The oral vasopressin receptor antagonist tolvaptan has been shown to be effective in adult patients with paraneoplastic SIADH, but data on broader use in cancer patients and the safety in pediatrics are not yet available (Kenz et al. 2011).
Cerebral Salt Wasting
Cerebral salt wasting (CSW) is a controversial diagnosis of unclear etiology; some authors suggest CSW does not exist or is exceedingly rare, while others suggest it is not cerebral in origin (Singh et al. 2002; Rivkees 2008). Nonetheless, CSW is a consideration in the pediatric neuro-oncology patient with hyponatremia and seizures. In reviewing children recovering from craniotomy for tumor resection, Hardesty et al. (2011) found that nearly half the patients with laboratory identified CSW developed postoperative seizures. The main clinical distinction between patients with SIADH and CSW is volume status; CSW represents hypovolemic hyponatremia, while patients with SIADH are either slightly hypervolemic or euvolemic. As in patients with SIADH, CSW patients will have hyponatremia, low serum osmolarity and relatively concentrated urine with high urine sodium excretion. The patient with CSW will often respond to isotonic fluid infusion, whereas normal saline frequently worsens hyponatremia in patients with SIADH. Correction of hypovolemia is the main therapy in CSW. Patients with chronic hyponatremia may require oral supplementation with salt tablets.
5.3.3.3 Infection
Patients receiving immunosuppressive chemotherapy have an increased risk for CNS infection which may present with AMS or seizure. Lumbar puncture is a helpful diagnostic tool in this setting, but should be deferred until the patient has been stabilized and CT imaging performed. Institution of broad antimicrobial coverage should not be delayed to obtain cerebrospinal fluid (CSF); cell counts can be informative in non-cytopenic patients even after sterilization of CSF and polymerase chain reaction (PCR)-based assays for viruses can remain positive after antiviral therapy has been instituted.
Infectious studies should include cultures of blood and CSF. Enterovirus, herpes simplex virus (HSV) and human herpesvirus-6 (HHV6) can be detected by CSF PCR. Culture or direct fluorescence antibody (DFA) testing of oropharyngeal lesions should also be considered. Treatment with high-dose acyclovir should be initiated urgently if viral meningoencephalitis is suspected.
A study of 40 cases of bacterial and fungal meningitis in pediatric oncology patients found that recent neurosurgery or a CNS device was present in the majority of cases (Sommers and Hawkins 1999). Among patients without neurosurgical intervention, those with hematologic malignancies and prolonged neutropenia were at the highest risk for infection.
5.3.3.4 Chemotherapy-Associated Neurotoxicity
Methotrexate, ifosfamide, and cytarabine (Ara-C) are commonly used pediatric chemotherapeutic agents that can provoke seizures and encephalopathy. Transverse myelitis is a peculiar complication described following the combined intravenous and intrathecal administration of cytarabine. Vincristine is most commonly known for its associated peripheral neuropathies, but medical errors in administration have highlighted its extreme neurotoxicity when introduced into the CNS or given at erroneously high doses systemically. Thiotepa, carmustine (BCNU) and busulfan are myeloablative agents used at high doses in stem cell transplant with neurologic dose-limiting toxicities (Papadopoulos et al. 1998). Other less commonly used agents such as nelarabine (a prometabolite of Ara-G) have similarly been implicated in patients developing new encephalopathy or seizures.
Methotrexate
Acute neurotoxicity has been described within 24 h of intravenous or intrathecal methotrexate administration, consisting of confusion, seizures, or encephalopathy. Methotrexate neurotoxicity is frequently self-resolving and is not related to drug levels (Rubnitz et al. 1998). By contrast, acute neurotoxicity following a methotrexate overdose can be very severe. Symptoms include myelopathy, seizures, encephalopathy and death from progressive necrotizing leukoencephalopathy. Published experience in overdose suggests benefit from high-dose intravenous (IV) leucovorin, IV carboxypeptidase G2 and alkalinization of the urine, with hemodialysis if renal insufficiency develops. If the overdose was administered intrathecally, the addition of CSF lavage or exchange and intrathecal administration of carboxypeptidase G2 have been used with good outcomes (Spiegel et al. 1984; Widemann et al. 2004).
Subacute methotrexate neurotoxicity includes a constellation of neurologic symptoms, often reversible, that occur after either IV or IT methotrexate. Symptoms include severe headache, focal or generalized seizures, AMS ranging from confusion to coma, and sensory and motor findings consistent with cerebrovascular accident. Aphasia is commonly reported (Dufourg et al. 2007). The median time from methotrexate administration to symptom development is consistently reported to be 10 days (Mahoney et al. 1998). The risk of developing methotrexate neurotoxicity increases with combination IV/IT therapy, increasing total cumulative administered methotrexate dose and cranial irradiation (Land et al. 1994; Reddick et al. 2005).
There is no single diagnostic test for methotrexate neurotoxicity; therefore recognizing the clinical scenario is vital to making the diagnosis. MRI can support the diagnosis (Fig. 5.2), as patients have characteristic changes on diffusion-weighted imaging that mimic ischemia (Reddick et al. 2005). However, unlike true ischemic changes, there is rapid normalization within several weeks (Sandoval et al. 2003). MRI findings are frequently multifocal and do not fit vascular distribution patterns. These MRI changes can also be seen in asymptomatic patients receiving methotrexate. Therefore, more extensive investigation to rule out other causes of seizure or AMS is often necessary.
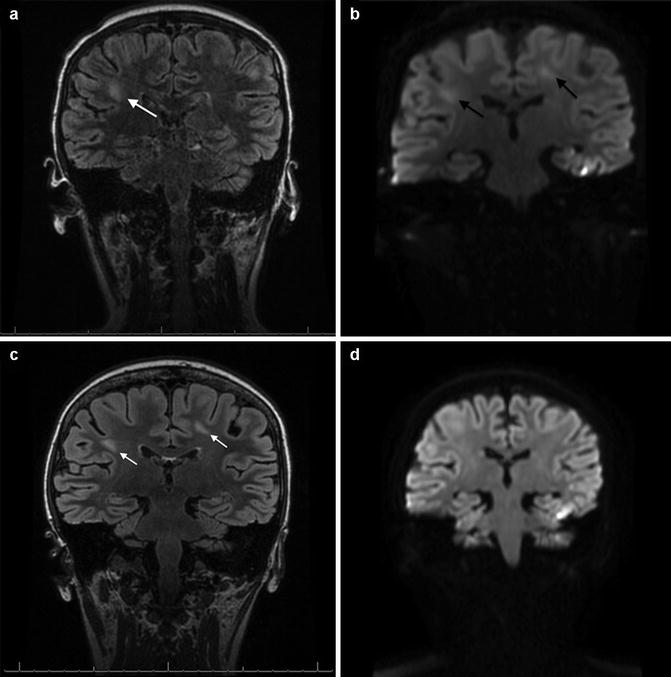
Fig. 5.2
Subacute methotrexate neurotoxicity. This 12-year-old patient with acute lymphoblastic leukemia developed symptoms of aphasia and lethargy approximately 10 days following combined intrathecal and high-dose intravenous (5 g/m2) methotrexate during delayed intensification. The T2-FLAIR MRI shown in Panel a demonstrates the development of ill-defined patches of hyperintensity within the white matter (white arrow). Diffusion-weighted imaging taken concurrently, shown in Panel b, shows restricted diffusion (black arrows). Although the pattern is not consistent with any one vascular territory, infarct is on the differential diagnosis with such findings. Fortunately, the patient’s symptoms resolved over 48 h, and while repeat scans taken 1 month later demonstrate continuation and evolution of the T2-FLAIR changes (Panel c, white arrows), the diffusion changes have now completely resolved (Panel d). The patient has continued to be completely asymptomatic but has not yet been rechallenged with intrathecal methotrexate
As mentioned, the majority of cases of methotrexate neurotoxicity are self-resolving. There are varying amounts of preclinical data and published case series that support the use of aminophylline or dextromethorphan in the acute phase and leucovorin rescue with future IT methotrexate in patients who have developed neurotoxicity in the past (Winick et al. 1992; Drachtman et al. 2002; Inaba et al. 2008). Aminophylline can be considered for patients who have acute obtundation after methotrexate. If toxicity is thought secondary to IT therapy, one can consider postponing the next scheduled IT methotrexate or substituting IT Ara-C and hydrocortisone, though there are very limited data to show this is an effective IT therapy in ALL. Patients can be safely rechallenged with IT methotrexate, either with or without leucovorin or aminophylline rescue, and not redevelop neurotoxicity (Rollins et al. 2004; Inaba et al. 2008). Thus, completely eliminating methotrexate from future therapy is not usually indicated.
Ifosfamide
Patients receiving ifosfamide may develop encephalopathy that can range from sleepiness and confusion to seizures or coma, starting several hours after infusion commencement. The reported frequency of this side effect is as high as 22 % in pediatric patients (Pratt et al. 1986). Although there is no clear dose-dependence, studies have suggested that hypoalbuminemia and renal dysfunction are risk factors (David and Picus 2005). The exact mechanism for ifosfamide-induced encephalopathy (IIE) is unclear although it is thought secondary to high levels of urine glutaric acid which is linked to one particular metabolite of ifosfamide, chloroethylamine (Kupfer et al. 1994). Methylene blue has been reported to successfully treat IIE and may also be effective as prophylaxis through unclear mechanisms, possibly by correcting derangement in mitochondrial flavoproteins (Pelgrims et al. 2000; Hamadani and Awan 2006). A review by Patel (2006) found that a lack of controlled evidence makes the effectiveness of methylene blue unclear. Thiamine has also been reported to be an effective treatment and prophylaxis for IIE through unclear mechanisms and also with a lack of controlled data (Hamadani and Awan 2006).
Cytarabine
The pyrimidine analog cytarabine is frequently used in the treatment of pediatric leukemia and lymphoma. There is a wide range of employed dosages and at lower doses neurologic toxicity is uncommon. Higher doses, however, are associated with an acute cerebellar syndrome in ≥10 % of patients (Baker et al. 1991). There is a clear dose-dependence of this side effect and it is generally seen at doses ≥3 g/m2/day; spacing administration from twice daily to once daily has been shown to reduce the incidence of cerebellar toxicity (Smith et al. 1997).
The clinical manifestations of cytarabine-induced cerebellar toxicity include the classic cerebellar signs of dysarthria, dysdiadochokinesia and ataxia (Smith et al. 1997). Cerebral dysfunction can also be seen in a subset of cases, with manifestations including encephalopathy, seizures and coma. Histopathologic changes seen in the cerebellar syndrome include loss of Purkinje cells in the cerebellar hemispheres and vermis along with a proliferation of glial cells known as Bergmann’s gliosis (Baker et al. 1991).
Symptoms often resolve following discontinuation of cytarabine; however some patients will have permanent neurologic damage. One large case series of adults and children receiving high-dose cytarabine found that approximately 1 % of patients who developed severe cerebellar symptoms had an irreversible or fatal course (Herzig et al. 1987). Although this same report demonstrated that some patients may be rechallenged without permanent neurologic damage, the decision to proceed with additional cytarabine is a difficult one. Identified risk factors for cerebellar syndrome include dose and timing as discussed above as well as hepatic or renal insufficiency and older age (Herzig et al. 1987; Baker et al. 1991; Rubin et al. 1992). No potential genetic modifiers have been reported in the literature.
IT cytarabine can be administered in its standard formulation or as a slow-release liposomal formulation (DepoCyt). The liposomal formulation appears to have a higher incidence of arachnoiditis; prophylactic corticosteroids may be beneficial in this situation (Glantz et al. 1999). As discussed, concomitant administration of IT and high-dose IV cytarabine is associated with the development of transverse myelitis presenting as bowel and bladder dysfunction with lower extremity weakness and should be avoided (Dunton et al. 1986; Schwenn et al. 1991).
5.3.3.5 Posterior Reversible Encephalopathy Syndrome
Posterior reversible encephalopathy syndrome (PRES, also called RPLS, reversible posterior leukoencephalopathy syndrome) was first described as a syndrome of encephalopathy accompanied by transient subcortical white matter changes on T2-weighted MRI (Hinchey et al. 1996). The clinical manifestations of PRES include headaches, seizures, AMS and cortical blindness. Hypertension is a clear antecedent and most significant risk factor in the development of PRES. Hypertension may be secondary to medications, such as corticosteroids, or concurrent medical conditions such as renal failure from tumor lysis syndrome (Greenwood et al. 2003). Retrospective studies also suggest a higher incidence of PRES during induction chemotherapy, independent of tumor lysis syndrome or hyperleukocytosis (Norman et al. 2007).
Since its initial description, PRES has been recognized to not be limited to the posterior circulation of the brain nor to white matter exclusively nor be fully reversible in all cases (Lucchini et al. 2008). While the initial report postulated a role for immunosuppression in the pathogenesis of PRES, it has been subsequently described in non-immunocompromised patients such as in pregnancy-induced hypertension. Specific immunosuppressive medications, such as tacrolimus and cyclosporine, continue to be implicated in case reports in part due to their effect on blood pressure.
PRES has been noted in pediatric malignancies including ALL, AML, non-Hodgkin lymphoma, and solid tumors including neuroblastoma, osteosarcoma, and Ewing sarcoma. Hypertension was a clearly identified preceding event in the majority of cases. Seizures were the most common clinical manifestation, with AMS, headache and some form of visual disturbance occurring in 30–40 % of cases (de Laat et al. 2011). While the majority of patients had a reversible course, 12 % had persistent neurologic symptoms.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


