- 17.1 Introduction
- 17.2 Infections
- 17.3 Demyelinating diseases of the central nervous system
- 17.3.1 Multiple sclerosis
- 17.4 Autoimmune diseases of the neuromuscular junction
- 17.4.1 Myasthenia gravis
- 17.4.2 Other autoimmune diseases of muscle end-plates
- 17.4.1 Myasthenia gravis
- 17.5 Immune-mediated neuropathies
- 17.5.1 Acute idiopathic polyneuritis (Guillain–Barré syndrome)
- 17.5.2 Chronic inflammatory peripheral neuropathies
- 17.5.1 Acute idiopathic polyneuritis (Guillain–Barré syndrome)
- 17.6 Paraneoplastic syndromes
- 17.7 Cerebral systemic lupus erythematosus
 Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
Visit the companion website at www.immunologyclinic.com to download cases with additional figures on these topics.
17.1 Introduction
The central and peripheral nervous systems are not excluded from immune disease. The immune system participates actively in nervous tissue to counteract infection, and so, as elsewhere, cells can enter these tissues in inflammation. T and B lymphocytes, as well as neutrophils, monocytes and macrophages, can invade inflamed nervous tissue. In infections and multiple sclerosis, intrathecal B cells are responsible for locally synthesized immunoglobulin found in the cerebrospinal fluid. T cells and macrophages, while protecting against infection, can also cause direct damage, as in chronic viral infection, post infective states and demyelination.
The blood–brain barrier normally excludes intravascular proteins (including IgG) and this must be breached before extrathecal circulating autoantibodies reach the central or peripheral nervous systems. In this way, circulating autoantibodies can be responsible for the pathogenesis of several antibody-mediated neuropathies, such as the Lambert–Eaton myasthenic syndrome (LEMS). This disease is an excellent example of the way in which an autoimmune pathogenesis has been proven by following clinical clues over many years (Box 17.1). Other autoimmune diseases selectively damage central or peripheral nervous tissue itself (e.g. multiple sclerosis and Guillain–Barré syndrome, respectively) or the muscle endplates (e.g. myasthenia gravis). On the other hand, involvement of nervous tissue may be part of a systemic disorder (e.g. in systemic lupus erythematosus).
| 1953 | Clinical description of disease |
| 1972 | LEMS associated with other organ-specific autoimmune diseases in patients- suggesting autoimmune cause |
| 1984 | Plasma exchange – successful therapy for affected patients suggesting pathogenic autoantibodies in plasma |
| 1987 | Human plasma from patients causes characteristic changes (electrophysiological + electron-microscopical) in mice proving humoral pathogenesis |
| 1991 | Autoantibodies to calcium channels detected in 20% of patients |
| 1991 | Autoantigen in small-cell cancer, present in 50% of LEMS patients |
| 1994 | Additional epitopes for antibodies to voltage-gated Ca2+ channels (VGCC) defined; now disease-specific antibodies detected in 100% of patients |
| 1995 | Randomized, placebo-controlled, cross-over study showed improved muscle strength with intravenous immunoglobulin therapy and associated reduction of autoantibdy titres (i.e. immunomodulation reduced pathogenic antibodies) |
| 1999 | Active immunisation with calcium channel peptides caused mild LEMS-like disease in rats. |
| 2006 | Passive transfer of disease from an affected mother to baby, causing transient neonatal weakness |
| 2007 | Mice transfected with mutated VGCC gene showed electrophysiological changes of LEMS |
17.2 Infections
The incidence of bacterial meningitis in infants has been reduced dramatically by means of immunization against the encapsulated organisms that are the major causes of these diseases. Normal children under the age of 2 years are unable to make antibodies to the carbohydrate capsules of Haemophilus influenzae type b (see Case 17.1), Streptococcus pneumoniae or Neisseria meningitidis and these pathogens accounted for 90% of meningitis seen in children until the early 1990s. Since the introduction of new vaccines against most of these pathogens, in which carbohydrate antigens have been coupled to protein carriers in order to provoke protective antibodies in infants and young children (see section 7.7), the incidence of meningitis due to Haemophilus influenzae type b and Neisseria meningitides type C has fallen dramatically in many countries (see Figure 7.8). However, routine immunization is only given at the age of 2 months and infants are susceptible until then (as in Case 17.1) if their mothers were not immunized. Now that these vaccines have been in use for >25 years, such infections in infants should now lead to suspicion of a primary immune deficiency in the mother. Disease in immunized children should also raise alarm bells!
 Case 17.1 Haemophilus influenzae type b meningitis
Case 17.1 Haemophilus influenzae type b meningitisAlice was a normal, full-term baby who was breast-fed and gained weight appropriately in the first 6 weeks of life. At 7 weeks she became acutely miserable, stopped feeding and her mother felt that she was very warm; when she took her temperature, it was 40°C. In the surgery, the doctor found that she had neck stiffness and Alice then vomited all over the couch. There was no rash or bruising but the left eardrum was inflamed. Meningitis was suspected; the doctor gave Alice an intramuscular injection of penicillin and instructed her mother to take her straight to the hospital where the on-call paediatrician was waiting. The clinical diagnosis of meningitis was confirmed and blood and cerebrospinal fluid (CSF) samples were taken immediately and intravenous antibiotics started. The CSF showed increased numbers of neutrophil leucocytes (131 × 106/l) and a few Gram-negative coccobacilli despite the initial dose of penicillin. Three days later these were shown to be Haemophilus influenzae and serotyping showed them to be Haemophilus influenzae type b. The full blood count showed a circulating neutrophilia (29 × 109/l), the C-reactive protein level was 230 mg/L. Alice made a rapid recovery with intravenous and subsequently oral antibiotics, with supportive management to ensure adequate ventilation and fluids. There were no long-term sequelae and she was immunised with childhood vaccines (including Hib) once she was fully recovered at 6 months (4 months later than healthy children).
Infections of the central nervous system (CNS), meningitis and encephalitis, are relatively uncommon in immunocompetent adults. Severe, unusual or recurrent brain infections should raise the possibility of an immune defect (see Chapter 3 and section 17.5), particularly an antibody or complement defect.
An other important example is human immunodeficiency virus (HIV) which leads to opportunistic microorganisms – viruses, fungi, parasites or intracellular bacteria – that infect the CNS (Table 17.1). The signs of inflammation in HIV-positive patients may be modified; fever and meningism are often absent and CSF may contain few cells, little protein and no detectable antibodies to the organisms involved, making diagnosis more difficult before the routine use of polymerase chain reaction (PCR) to detect infective agents directly.
Table 17.1 Important neurological complications of human immunodeficiency virus (HIV) infection
| Primary HIV infection Dementia Atypical meningitis Myelopathy |
| Opportunistic infections Cerebral toxoplasmosis Meningitis – cryptococcal, tuberculous Encephalitis due to cytomegalovirus/herpes simplex virus Progressive multifocal leucoencephalopathy (papovavirus) Cytomegalovirus retinitis |
| Tumours associated with viruses Kaposi’s sarcoma [HHV-8] B-cell lymphoma [EBV] |
In addition, the causative virus, HIV, can itself infect the brain to produce a range of problems including AIDS dementia. Neurological abnormalities occur clinically in about 50% of HIV-infected adult patients and in many HIV-infected children. Subclinical disease may be even more common, since up to 75% of brains of AIDS patients are found to be affected post-mortem. Pathological changes range from white matter pallor and mild lymphocytic infiltration to macrophage abnormalities (including multinucleate giant cells) associated with macrophage activation. Computed tomography (CT) may show cerebral atrophy.
Infections can result in damage even after the causative pathogen has been eliminated. Parainfectious encephalitis occurs some time following a childhood viral illness (rubella, measles, chicken pox). This is due to demyelination following bystander activation of T cells or molecular mimicry resulting in specific T cells mistaking myelin for virus (see section 5.5). Although the condition is usually self-limiting, permanent damage or death may result.
In contrast, subacute sclerosing panencephalitis (SSPE) is a rare, progressive disease of children, who present with insidious dementia. The disease follows measles infection several years earlier, and high levels of specific anti-measles antibody as well as measles virus are found in the brain, blood and CSF. The intrathecal IgG is not only oligoclonal but also can be absorbed out with measles virus itself. There is no obvious defect in the adaptive immune responses to measles virus; such children may have defective innate immunity, in particular abnormal TLR receptors that are important in early protection against many viruses.
Progressive multifocal leucoencephalopathy is a rare demyelinating disease that can be induced by papovaviruses and typically occurs in immunosuppressed patients, such as those with AIDS or those receiving immunosuppressive treatment, including therapeutic monoclonal antibodies, particularly Natalizumab, which blocks anti-α4-integrins required for migration of leucocytes that would control the papovavirus.
17.3 Demyelinating diseases of the central nervous system
17.3.1 Multiple sclerosis
Multiple sclerosis (MS) is common in Caucasians, with a prevalence of 1 case per 2000 population (see Table 3.1 in Chapter 3). It is an inflammatory disease of white matter in the CNS. It is a hugely variable disease, in terms of disability, rate of progression and prognosis. The clinical diagnosis of MS can be difficult, as definite histopathological confirmation by biopsy is not done in life. The clinical features depend on the site of the pathological lesion in the brain (as in Case 17.2); many plaques are clinically silent. The rate of new lesions is unpredictable and also variable. Subgroups of MS depend on the rate of progression and recovery between attacks; relapsing and remitting MS is quite distinct from progressive disease.
 Case 17.2 Multiple sclerosis
Case 17.2 Multiple sclerosisA 38-year-old woman presented with tingling, numbness and clumsiness of both hands for 1 week, with a band of numbness from the umbilicus to the axillae. Six months earlier, following an upper respiratory tract infection, she had experienced a short episode of blurred vision that she put down to tiredness. She was now anxious because her maternal grandmother had suffered from multiple sclerosis (MS).
On neurological examination, she had absent abdominal reflexes with brisk tendon jerks and bilateral extensor plantar responses. Blood investigations were normal, including a full blood count, C-reactive protein, vitamin B12 and folate levels and syphilis serology. A lumbar puncture was carried out. The cerebrospinal fluid (CSF) investigation results are shown in Table 17.2. Oligoclonal IgG bands (see Fig. 19.8) are not found in normal CSF, but are found in 90% of patients with MS; in the absence of clinical signs of infection, this test is almost diagnostic of MS (see Box 17.2).
Table 17.2 Cerebrospinal fluid investigations in Case 17.2, multiple sclerosis
| Protein concentration | 0.4 g/L (NR 0–0.4 g/L) |
| Red blood cells | None |
| Lymphocytes | 3 × 106/L (NR < 5 × 106/L) |
| IgG/albumin ratio | 26% (NR 4–22%) |
| Isoelectric focusing | Oligoclonal bands present |
The clinical diagnosis was multiple sclerosis; other possible diagnoses, such as neurosyphilis or subacute combined degeneration of the cord, were excluded by investigation. MRI showed some silent lesions; she asked to be followed carefully without specific therapy at this stage in view of her mild symptoms.
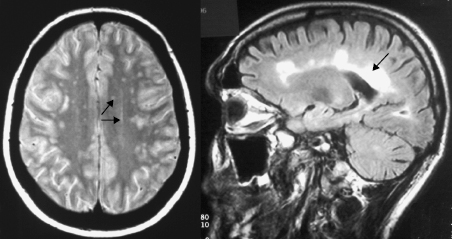
Optic neuritis is a similar inflammatory, demyelinating disease of one or both optic nerves, with recovery in 75–90% of patients. An oligoclonal pattern of IgG is found in CSF from most patients with optic neuritis and nuclear magnetic resonance imaging (MRI) shows silent lesions elsewhere in the brain. It can be due to many causes, but approximately 50–80% of these patients will develop MS within 15 years, as in Case 17.2. Clinical diagnosis of MS depends on evidence of at least two attacks separated in time and sites of lesions, with exclusion of all other causes, as in Case 17.2. Confirmatory tests are required. Poor nerve conduction can be demonstrated by prolonged responses on evoked potential testing. MRI of the brain (Fig. 17.1) shows lesions in 90% of affected individuals but, like all imaging, is non-specific and can be unreliable, especially in elderly people. International protocols are available to neuroradiologists to standardize their interpretations of MRI scans in these patients.
Fig. 17.1 MRI of brain of MS patient showing white matter changes due to demyelination (arrows). Axford J & O’Callaghan C (eds) Medicine, 2e (2004). Reproduced with permission of John Wiley & Sons Ltd.
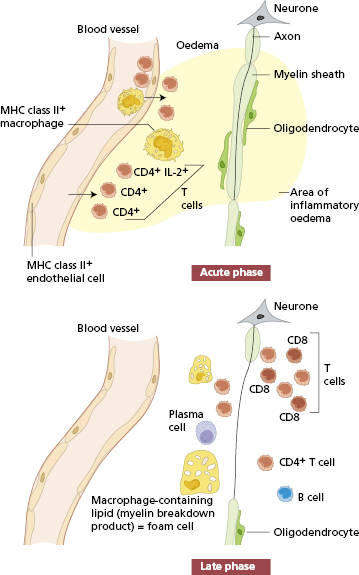
The pathological lesion in MS, seen at post-mortem brain biopsy, is a ‘plaque’; this is an area of the white matter in which myelin and oligodendrocytes are absent. Myelin is a protein–phospholipid material that surrounds axons in a multilayered, dense spiral. Myelin sheaths in the CNS are formed by compacted membranes of oligodendrocyte extensions. The integrity of the myelin sheaths depends on the maintenance of normal oligodendrocyte function. Axons without myelin sheaths are poor conductors of nerve impulses, resulting in a neurological deficit. In the early stages of a plaque, there is tissue oedema, apoptotic oligodendrocytes and infiltrating cells; lymphocytes and macrophages are seen around the venules in the area. B cells are involved in the local production of immunoglobulin, while others, including T cells and macrophages, are part of the acute inflammatory process (Fig. 17.2).
Routine examination of CSF is not enough. It is essential to look at the nature of the immunoglobulin (Box 17.2
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


