56 Neuroblastoma
Epidemiology, Screening, Etiology, and Genetics
Neuroblastoma is the most common extracranial solid tumor in children and the most common malignancy of infants. Approximately 650 new cases are diagnosed annually in the United States, with an incidence of 0.9 per 100,000.1 The tumor accounts for 8% to 10% of all childhood cancers and is slightly more common in boys than in girls, with a male-to-female ratio of 1.1 : 1. Review of more than 3000 patients registered on studies at Pediatric Oncology Group (POG) institutions from 1990 to 2000 and at Children’s Cancer Group (CCG) institutions from 1991 to 1995 show the median age at diagnosis is 17.3 months; 40.1% are younger than 1 year of age, 89.4% are younger than 5 years of age, and 97.8% are younger than 10 years of age.2
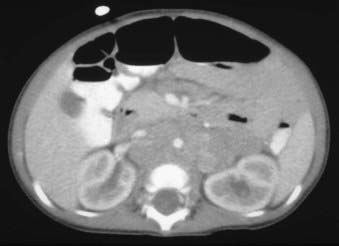
Neuroblastoma arises from primitive adrenergic neuroblasts of neural crest tissue. This tissue forms columns that are the precursors of spinal ganglia, dorsal spinal nerve roots, and chromaffin cells of the adrenal medulla. Thus, the majority of neuroblastoma cases occur in an anatomic distribution consistent with the location of neural crest tissue. The location of tumors at the time of diagnosis varies with age (Table 56-1). The adrenal gland is the most common location for the development of neuroblastoma, accounting for 35% of cases (Figure 56-1). Paraspinal ganglia in the low thoracic and abdominal chains constitute 30% of all cases, the posterior mediastinum accounts for 19%, and pelvic and cervical ganglia account for 2% or fewer.3 It has been reported that microscopic neuroblastic nodules, resembling neuroblastoma in situ, were found frequently at autopsy in infants younger than 3 months of age who died of unrelated causes.4 Initially, this was believed to mean the incidence of neuroblastoma was much higher during early infancy, but these tumors regressed spontaneously and were never clinically detected. However, studies have demonstrated that these nodules occur uniformly in all fetuses, peak between 17 and 20 weeks of gestation, and gradually regress by the time of birth or shortly thereafter.5 Thus, the development and subsequent regression of neuroblastoma nodules most likely represents a normal embryologic event, and the development of clinically detectable neuroblastoma is a consequence of a disruption of this process.
Screening Studies
The most extensive data regarding neuroblastoma screening come from Japan, Germany, and a combined Canadian and U.S. study. Neuroblastoma screening has been in place in Japan since 1981. During a 12-year screening period, the incidence rate in infants younger than 1 year of age rose from 28 to 260 per million, but there was no reduction in the incidence rate in children older than 1 year of age.6 The Japanese have also noted a reduction in overall mortality since the inception of mass screening.7 However, this reduced mortality may reflect the improving treatments that have arisen during the same time and not be a true reflection of the benefits of screening. In Canada, they have also seen an increase in the incidence of early stage disease in children less than 1 year of age.8 There has been no reduction in the incidence of advanced stage disease in children older than 1 year of age or in the mortality from neuroblastoma based on population screening at 3 weeks and 6 months of age in Canada.9 Screening at 1 year of age in Germany also did not reduce the incidence of advanced-stage disease in children or the mortality of children diagnosed with neuroblastoma.10 In addition, the psychological affect of false-positive tests and the potential risk incurred by interventions performed on clinically insignificant tumors has not been fully quantified, but could be significant. Many of the tumors detected by mass screening have now been observed, and more than one-third spontaneously regress.11 Thus the benefits of mass screening seem limited and do not appear to warrant the costs.
Genetics
The cause of neuroblastoma is unknown in most cases. No prenatal or postnatal exposure to drugs, chemicals, or radiation has unequivocally demonstrated an increased incidence of neuroblastoma.12,13 Genetics, however, play an extremely important role. A case control study in France (ESCALE) showed a positive association between congenital malformation and neuroblastoma, particularly in infants less than one year of age.14 Familial neuroblastoma is also well described and presents at an earlier median age (9 months versus 22 months for sporadic cases). At least 20% of patients with familial neuroblastoma have bilateral or multifocal disease.15 Genetic studies of hereditary disease have been impeded by the rarity of the condition and the small size of pedigrees caused by the lethality of neuroblastoma in early childhood. A family history of neuroblastoma is obtained in only approximately 1% of patients. Studies of such families suggest locus heterogeneity; no commonly mutated gene has been identified, and the penetrance appears to be low.16 Genetic loci in different families include germline mutations in PHOX2B, seen in patients with associated abnormalities such as congenital hypoventilation syndrome or Hirschsprung disease, and abnormalities of the short arm of 16.17–20 Neuroblastoma has also been seen occasionally in patients with constitutional chromosomal rearrangements, including deletions overlapping putative tumor suppressor loci at chromosome bands 1p36 and 11q14-23.21–23 Thus neuroblastoma predisposition is genetically heterogeneous, and initiation of tumorigenesis may require multiple alterations. A recent study using whole-genome single-nucleotide polymorphism analysis revealed a common genetic variation at chromosome band 6p22 that is associated with susceptibility to neuroblastoma, particularly with high-risk disease.24 Very recently, a number of studies have appeared showing that germline mutations or amplifications in the ALK gene are found in approximately 10% of sporadic neuroblastoma and germline mutations in the majority of heritable neuroblastoma.25
Deletion of the distal short arm of chromosome 1 (1p) was the first described genetic mutation in neuroblastoma tumors and is the most consistently reported abnormality, occurring in 70% to 80% of the karyotyped near-diploid tumors.26 This most likely represents a deletion (and loss of heterozygosity [LOH]) of a tumor suppressor gene and is found more commonly in advanced disease. Homogeneous staining regions (HSRs) and double-minute chromatin bodies (dmins) are a manifestation of gene amplification. Both of these are derived from the distal short arm of chromosome 2 (2p), which contains the proto-oncogene MYCN.27
MYCN amplification occurs in roughly 25% of primary neuroblastoma cases, 5% to 10% in patients with low stages of disease and stage IV-S, and 30% to 40% in patients with advanced disease.28 The MYCN proto-oncogene is derived from the c-myc viral oncogene and, as mentioned previously, is located on 2p, thus correlating with the cytogenetic abnormalities of HSRs and dmins. If it is going to occur, MYCN amplification is almost always present by the time of diagnosis. This suggests that it is an intrinsic property of a subset of aggressive tumors with a poor prognosis.27 The development of an overexpressing TH-MYCN transgenic mouse that spontanously develops neuroblastoma is further evidence of a role of this oncogene in tumorigenesis.29 Molecular studies have shown a strong correlation between 1p LOH and MYCN, suggesting these two genetic events may be related.26
Chromosomal ploidy is another marker of prognosis that is particularly useful in infants younger than 18 months of age. Near-diploid or pseudodiploid tumors have near-normal nuclear DNA content but often have structural chromosomal abnormalities, including MYCN amplification. Hyperdiploid or near-triploid tumors typically lack MYCN amplification and 1p deletion and therefore have an excellent prognosis.30,31
Telomerase activity has been correlated with MYCN amplification and poor outcomes while increased expression of high-affinity nerve growth factor receptor (gp140TRK-A) has been associated with favorable outcomes. Conversely, the lack of expression of gp140TRK-A has been associated with MYCN amplification and poor survival.27
Pathologic Conditions
Neuroblastoma is a classic “small round blue cell” tumor. Others include Ewing sarcoma, non-Hodgkin lymphoma, primitive neuroectodermal tumors, and rhabdomyosarcoma. Histologic subtypes, including neuroblastoma, ganglioneuroblastoma, and ganglioneuroma, represent different points along the maturation pathway in order of increasing differentiation. The typical neuroblastoma is composed of small, uniform cells containing dense, hyperchromatic nuclei and scant cytoplasm. The presence of neuritic processes (neuropil) is pathognomonic. Homer-Wright pseudorosettes, neuroblasts surrounding areas of eosinophilic neuropil, are seen in 15% to 50% of cases. Ganglioneuromas are composed primarily of mature ganglion cells, neuropil, and Schwann cells, and behave in a benign fashion. Ganglioneuroblastomas have histopathologic characteristics of both neuroblastomas and ganglioneuromas. Because histopathologic features may vary within any single tumor, multiple sections must be examined.32,33 Distinguishing neuroblastomas from other small round blue cell tumors requires special techniques including immunohistochemistry and electron microscopy. Neuroblastoma stains with monoclonal antibodies that recognize neuron-specific enolase, synaptophysin, and neurofilament. Electron microscopy reveals neurosecretory granules that contain catecholamines, microfilaments, and parallel arrays of microtubules within the neuropil.34
Although many different classification systems have been used to help define the prognosis for neuroblastoma, the International Neuroblastoma Pathology Commmittee (INPC) defined and tested a modification of the Shimada system, now widely accepted and validated.32 It is divided into favorable and unfavorable prognostic groups based on histologic category, age, amount of Schwann cell stroma, degree of differentiation; and the mitosis-karyorrhexis index. Each of these features are also independently prognostic.35 In addition, nodular versus diffuse histologic pattern is noting macro nodules, which tend to be associated with a poor prognosis compared with intermixed nodules. Characteristics of the INPC system are listed in Table 56-2.
Clinical Presentation
As mentioned previously, primary tumor location varies with age (see Table 56-1). Extent of disease is also age-dependent; the majority of children younger than 1 year of age have localized disease at the time of diagnosis, whereas the majority of children older than 1 year of age have disseminated disease (Table 56-3). Signs and symptoms of neuroblastoma depend on location of the primary tumor but can also reflect lymph node status and metastatic spread. Bone marrow, bone, liver, and skin are the most common sites for hematogenous spread. Rarely, disease spreads to the lungs and brain. Table 56-4 lists the associated signs and symptoms for each anatomic area of disease. Because of the type of catecholamines secreted from most neuroblastomas, hypertension, flushing, and tachycardia are uncommon symptoms. Other rare paraneoplastic symptoms are secretory diarrhea, resulting from secretion of vasoactive intestinal peptide,36 and opsoclonus-myoclonus ataxia syndrome, most likely resulting from antineuronal antibodies that cross-react with cerebellar tissue.37
Table 56-4 Signs and Symptoms of Disease Based on Anatomic Location of Primary Tumor and Metastases
| Primary Site | Signs and Symptoms |
|---|---|
| Abdomen | |
| Cervical | |
| Thoracic | |
| Pelvis | |
| Paraspinal location in cervical, abdominal, thoracic, or pelvic areas | |
| Metastatic Disease | |
| General | |
| Liver | |
| Bone | |
| Skin | |
| Bone marrow | |
| Brain | |
| Paraneoplastic Syndromes | |
• Opsoclonus-myoclonus-truncal ataxia syndrome (myoclonic jerking and random eye movements); associated with early stage neuroblastoma and may persist after cure |
Diagnostic and Staging Studies
In terms of regional and distant disease, CT and MRI can assess nodal disease, hepatic metastases, and intraspinal extension. CT and MRI are also useful in determining whether metastases to the skull, orbit, mandible, or brain are present. Conventional radiographs may be used to assess painful bone metastases, but the standard and most sensitive agent for detection of osteomedullary involvement is 123I-metaiodobenzylguanidine (MIBG) scan, which concentrates as well in primary tumors and soft tissue metastases. MIBG is a guanethidine derivative and an analogue of norepinephrine, and therefore specifically taken up and stored in tumors of sympathetic origin, which express the norepinephrine transporter.38 Because of the high specificity and sensitivity in neuroblastoma, 123I-MIBG has superseded the use of technetium bone scans for the detection of skeletal metastases in the majority of children with neuroblastoma tumors, which take up the chemical in greater than 90% of cases, and has been recommended by the last international consensus conference as a standard element of staging and response evaluation.39,40 A 99mTc bone scan is indicated for detection of bone metastases in the occasional patient whose tumor does not take up MIBG. 18F-deoxyglucose positron emission tomography is also a useful complementary imaging modality that reflects the metabolic activity of primary and metastatic lesions, but its exact role in staging and response is still under investigation.41,42 Metastatic lesions are most often located in the periorbital region, in the metaphyses of long bones, and in the axial skeleton. Complete staging includes two bilateral posterior iliac crest bone marrow aspirates and biopsies. A single positive result is sufficient for the documentation of bone marrow involvement.39
Excess catecholamines are produced in nearly 90% of cases. Therefore, part of the neuroblastoma workup includes measuring urine catecholamines and their metabolites, specifically norepinephrine, vanillylmandelic acid, 3-methoxy-4-hydroxylphenylglycol, or homovanillic acid. Urine or serum dopamine may be measured. Urinary catecholamines are often given as ratios to urinary creatinine.43
According to the international criteria for diagnosing, staging, and assessing response to treatment, the diagnosis of neuroblastoma is established under one of the following circumstances: (1) An unequivocal pathologic diagnosis is made from tumor tissue by light microscopy (with or without immunohistology, electron microscopy, increased urine or serum catecholamines or metabolites), or (2) Bone marrow aspirate or trephine biopsy contains unequivocal tumor cells and urine or serum catecholamines or metabolites are increased.39
Staging and Prognostic Factors
Several different staging systems have been used in neuroblastoma. The Evans and D’Angio classification, historically used by the CCG classification, is the original system. It is based on the extent of the primary tumor and the presence or absence of distant disease. It includes stages I, II, III, IV, and IV-S.44 The POG used an alternative staging system that included initial resectability of the primary and lymph node status, although the importance of lymph node involvement in neuroblastoma is unclear. It includes stages A, B, C, D, and D-S.45,46 The differences between these two systems can be substantial, depending on the stage. Therefore, a group of investigators met at two international conferences to arrive at a consensus regarding neuroblastoma staging. Criteria for diagnosing, staging, and assessing response to treatment was set forth, and the International Neuroblastoma Staging System (INSS) is now generally accepted as the standard staging system (Table 56-5). To interpret the literature, it is important to recognize the similarities between the INSS and the other two major staging systems. Stage 1 is similar to stages I and A. Stage 4 is essentially the same as stages IV and D, and stage 4S is the same as IV-S and D-S. The greatest disagreement concerns the middle stages (II and III; B and C). Therefore, the INSS divided stage 2 into 2A (incompletely excised tumor) and 2B (ipsilateral lymph node involvement) to evaluate whether patients with 2B disease behave more like stage 2A patients or stage 3 patients. In addition to revising staging, the INSS delineated standard definitions of response to treatment (Table 56-6). These revisions and standard definitions were designed to make interpreting the literature and making management decisions uniform.39
Table 56-5 International Neuroblastoma Staging System
* The midline is defined as the vertebral column. Tumors originating on one side and crossing the midline must infiltrate to or beyond the opposite side of the vertebral column.
† Marrow involvement in stage 4S should be minimal (i.e., <10% of total nucleated cells identified as malignant on bone marrow biopsy or on marrow aspirate). More extensive marrow involvement is considered to be stage 4. The metaiodobenzylguanidine scan (if performed) should be negative in the marrow.
From Brodeur GM, Seeger RC, Barrett A, et al: International criteria for diagnosis, staging and response to treatment in patients with neuroblastoma, J Clin Oncol 6:1874–1881, 1988; and Brodeur GM, Pritchard J, Berthold F, et al: Revisions in the international criteria neuroblastoma diagnosis, staging and response to treatment, J Clin Oncol 11:1466–1477, 1993.
Table 56-6 INSS Classification of Responses to Therapy
| Response | Metastatic Sites/Markers | Primary Tumor |
|---|---|---|
| Complete response (CR) | No tumor | No tumor; catecholamines normal |
| Very good partial response (VGPR) | Decreased by 90%-99% | No tumor; catecholamines normal; residual 99Tc |
| Partial response (PR) | Decreased by >50% | All measurable sites decreased by >50% |
| Bones and bone marrow | Number of positive bone sites decreased by >50%; no more than one positive bone marrow site allowed* | |
| Mixed response (MR) | No new lesions | >50% reduction of any measurable lesion (primary or metastases) with <50% reduction in any other; <25% increase in any existing lesion |
| No response (NR) | No new lesions | <50% reduction but <25% increase in any existing lesion |
| Progressive disease (PD) | Any new lesion | Increase of any measurable lesion by >25%; previous negative marrow biopsy positive for tumor |
INSS, International Neuroblastoma Staging System.
* One positive marrow aspirate or biopsy allowed for PR if this response represents a decrease from the number of positive sites at diagnosis.
From Brodeur GM, Pritchard J, Berthold F et al: Revisions in the international criteria neuroblastoma diagnosis, staging and response to treatment, J Clin Oncol 11:1466–1477, 1993.
A more recent international consensus conference was held in 2005 to further refine the staging and risk classification for neuroblastoma. This International Neuroblastoma Risk Group (INRG) Task Force proposed a simpler staging, based on whether or not a tumor had image-defined risk factors for surgery (L1: no risk factors; L2: risk factors present),47 and whether or not there was metastatic disease.48 In addition, further refinements were made to the response criteria, to incorporate semiquantitative scoring for the MIBG scan49,50 and standardization of bone marrow disease using immunocytology and real time–polymerase chain reaction.51,52
Treatment for neuroblastoma is based on risk stratification. Therefore, it is essential to understand the clinical and biologic variables predictive of disease outcome. The most important clinical risk factors are disease stage and age at diagnosis. Infants younger than 18 months have a more favorable outcome compared with older children with the same disease stage, as shown in a retrospective analysis of 3666 patients (1986 to 2001) from patients in the Children’s Oncology Group (COG) with documented follow-up data. Using a cut-off of 460 days of age, the 4-year event-free survival (EFS) for those younger than 460 days was 82% versus 42% for those older than 460 days (P < 0.0001); using a cut-off of 573 days, the 4-year EFS was 74% and 38%, respectively (P < 0.0001). The 18-month (547-day) cut off was chosen for convenience, because the difference in EFS was slight over the range of 15 to 18 months. MYCN gene amplification and stage 4 were also more frequent in the older patients, but even after adjustment for these factors, age remained significant.53 Similarly, several biologic variables have prognostic significance. A new histopathologic classification, the INPC, based on the original Shimada system, is now the international standard. Like the Shimada system, this classifies patients into favorable and unfavorable groups based on age, amount of Schwann cell stroma, degree of differentiation, and mitotic-karyorrhectic index (see Table 56-2). Further analyses of an international database of 8800 neuroblastoma patients showed that each of the components of the INPC had independent prognostic significnace within a given age group.35 Serum markers including ferritin, neuron-specific enolase, lactate dehydrogenase, and GD2 each have prognostic value (increased levels associated with worse prognosis); however, they are not currently used in risk stratification because their multivariate importance is less significant than other factors. Unfavorable genetic prognostic markers that are most significant in multivariable analysis include MYCN gene amplification, diploidy, and11q LOH. Other genetic changes with prognostic value in univariate analysis include 1p LOH, TRKA expression, and 17q additions, though these are not currently used in risk stratification.35
Standard Therapeutic Approaches
Surgery
Surgery is used to establish the diagnosis, provide tissue for biologic studies, determine stage, establish vascular access for chemotherapy, and accomplish therapeutic excision of the primary tumor and involved regional lymph nodes. The operative approach depends on the initial risk evaluation, which in turn is a function of tumor location, mobility, relationship to major vessels and nerves, ability to control blood supply, presence of distant metastases, and overall prognosis. More recently, image-defined risk factors have been used to determine resectability.54 These risk factors are based in the main on vascular or neural encasement as determined by imaging studies. In patients with high-risk disease, initial surgery is directed toward diagnosis, staging, vascular access, and adequate tissue sampling for diagnostic studies. Biopsy of the liver at the time of initial surgery is proposed for patients with abdominal tumors without clinical evidence of metastatic disease.2
Nonadherent, intracavitary lymph nodes should be sampled, including lymph nodes superior and inferior to the primary tumor. Lymph nodes adherent to the primary tumor have little relevance in predicting outcome but should be removed if grossly involved. High-risk patients always receive neoadjuvant chemotherapy prior to primary site resection as this increases the chance of complete removal and reduces the risk of complications, especially nephrectomy.55 In high-risk patients, gross total resection of the primary tumor site and involved regional lymphatics seems to be associated with improved outcome, and especially improved local control (LC).56–57
Radiation Therapy
Neuroblastoma is a relatively radiosensitive tumor. Data collected on neuroblastoma cell line show a D0 of 1.04 Gy and an n of 1.36, which is much more radiosensitive than other mammalian tumors.58 However, despite its radiosensitivity in the laboratory, its clinical response to irradiation is variable, which may be attributed to its variation in genomic amplification of MYCN.59

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


