Myeloproliferative Neoplasms and Myelodysplastic Syndromes
Jad Chahoud
Srdan Verstovsek
Guillermo Garcia-Manero
Hagop M. Kantarjian
Elias Jabbour
I. BCR-ABL1-NEGATIVE MYELOPROLIFERATIVE NEOPLASMS
A. Overview
The myeloproliferative neoplasms (MPNs) are clonal disorders of pluripotent hematopoietic stem cells or of lineage-committed progenitor cells. MPNs are characterized by autonomous and sustained overproduction of morphologically and functionally mature granulocytes, erythrocytes, or platelets. Although one cellular element is most strikingly increased, it is not uncommon to have modest or even major elevations in other myeloid elements (e.g., thrombocytosis and leukocytosis in patients with polycythemia vera [PV]). Bone marrow (BM) aspirates and biopsy specimens typically show hyperplasia of all myeloid lineages (panmyelosis). Morphologic maturation and cellular function are essentially normal, although platelet dysfunction occasionally contributes to bleeding. The overproduction of blood elements in MPNs now appears related to “switched-on” tyrosine kinase signaling pathways. For chronic myelogenous leukemia (see Chapter 19), this arises from the t(9;22) translocation and the BCR-ABL1 gene product. On the other hand for the BCR-ABL1-negative MPNs, a single nucleotide mutation in the gene coding for JAK2, a tyrosine kinase normally activated by erythropoietin and other cytokines, plays an analogous role. This group of MPNs that encompasses PV, essential thrombocythemia (ET), both primary and secondary myelofibrosis (MF),1 is discussed in the first section of this chapter. The incidence of PV ranges from 0.4 to 2.8/100,000/year, ET 0.4 to 3.4/100,000/year, and primary myelofibrosis (PMF) 0.8 to 2.1/100,000/year.
Ever since the observation of William Dameshek in 1951, that PV, ET, and PMF were a set of closely interrelated disorders, the understanding of the pathogenesis of classical MPNs has come a long way. This evolution has been marked by three major milestones that rendered MPNs among the best genetically characterized malignancies. The first finding was in 2005, with the detection of somatically acquired mutations of the gene JAK2 in the majority of patients with MPNs. The point mutation V617F in exon 14 of JAK2 is present in 74% to 97% of patients with PV, and in 30% to 65%
of patients with ET or PMF. The second breakthrough occurred in 2006, with the detection of the myeloproliferative leukemia (MPL) virus oncogene mutations in 3% to 5% and 8% to 11% of nonmutated JAK2 ET and PMF, respectively. Lastly, the third milestone was in 2013, when mutations in calreticulin (CALR) gene were observed in approximately 50% to 70% of patients with ET or PMF who do not carry a mutation in either the JAK2 or MPL gene.2 The above-mentioned genetic discoveries have rendered approximately 90% of patients with MPN carriers of a mutually exclusive mutation in only one of these three genes: JAK2, MPL, or CALR.3 Therefore, positivity for JAK2 V617F gives important diagnostic confirmation for MPNs, although a negative result is not basis for exclusion. Other discovered mutations are less specific to MPNs but are prognostically relevant, such as additional sex combs-like 1 (ASXL1) mutation. The prognostic value varies by mutation and can be summarized as follows; in ET, JAK2 mutations are associated with increased risk of thrombosis, while in PMF patients, type 1 or type 1-like CALR mutations are associated with superior survival, and ASXL1 mutations with inferior survival.3,4 Major MPN-related complications comprise thrombosis and leukemic or fibrotic transformation. The management of MPNs was also revolutionized with the discovery of these new mutations through the development of molecularly targeted therapy, including JAK inhibitors. JAK inhibitors have shown promising activity in controlling splenomegaly and constitutional symptoms in PMF and PV, which led to the recent approval of the JAK inhibitor ruxolitinib for use in hydroxyurea-resistant PV. In view of these changes, the next sections discuss the diagnosis, evolution, prognosis, and the recent therapeutic advances for PV, ET, and PMF.
of patients with ET or PMF. The second breakthrough occurred in 2006, with the detection of the myeloproliferative leukemia (MPL) virus oncogene mutations in 3% to 5% and 8% to 11% of nonmutated JAK2 ET and PMF, respectively. Lastly, the third milestone was in 2013, when mutations in calreticulin (CALR) gene were observed in approximately 50% to 70% of patients with ET or PMF who do not carry a mutation in either the JAK2 or MPL gene.2 The above-mentioned genetic discoveries have rendered approximately 90% of patients with MPN carriers of a mutually exclusive mutation in only one of these three genes: JAK2, MPL, or CALR.3 Therefore, positivity for JAK2 V617F gives important diagnostic confirmation for MPNs, although a negative result is not basis for exclusion. Other discovered mutations are less specific to MPNs but are prognostically relevant, such as additional sex combs-like 1 (ASXL1) mutation. The prognostic value varies by mutation and can be summarized as follows; in ET, JAK2 mutations are associated with increased risk of thrombosis, while in PMF patients, type 1 or type 1-like CALR mutations are associated with superior survival, and ASXL1 mutations with inferior survival.3,4 Major MPN-related complications comprise thrombosis and leukemic or fibrotic transformation. The management of MPNs was also revolutionized with the discovery of these new mutations through the development of molecularly targeted therapy, including JAK inhibitors. JAK inhibitors have shown promising activity in controlling splenomegaly and constitutional symptoms in PMF and PV, which led to the recent approval of the JAK inhibitor ruxolitinib for use in hydroxyurea-resistant PV. In view of these changes, the next sections discuss the diagnosis, evolution, prognosis, and the recent therapeutic advances for PV, ET, and PMF.
B. Polycythemia vera
1. Diagnosis
In the golden era of disease description, around the end of the 19th century, Vaquez was the first to describe the idiopathic entity, PV (maladie de Vaquez). Later on, Osler established it as a new clinical entity distinguished from secondary and relative polycythemia.
PV must be distinguished from relative or spurious polycythemia (normal red blood cell [RBC] mass, decreased plasma volume) and from secondary erythrocytosis (increased RBC mass due to hypoxia, carboxyhemoglobinemia, inappropriate erythropoietin syndromes with tumors or renal disease, etc.). PV is suspected in patients with hemoglobin levels greater than 18.5 g/dL in men or 16.5 g/dL in women or hemoglobin levels greater than 17 g/dL in men or 15 g/dL in women if associated with a documented and sustained increase of at least 2 g/dL from an individual’s baseline value. Diagnostic evaluation begins with peripheral JAK2 V617F mutation screen and measurement of
serum erythropoietin levels. This is because JAK2 is present in 97% of patients with PV and is not associated with other causes of increased hemoglobin/hematocrit levels; similarly, a subnormal serum erythropoietin level is expected and encountered in more than 90% of patients with PV but not in secondary or apparent polycythemia. However, neither the absence of JAK2 nor the presence of a normal erythropoietin level rules out the diagnosis of PV.3
serum erythropoietin levels. This is because JAK2 is present in 97% of patients with PV and is not associated with other causes of increased hemoglobin/hematocrit levels; similarly, a subnormal serum erythropoietin level is expected and encountered in more than 90% of patients with PV but not in secondary or apparent polycythemia. However, neither the absence of JAK2 nor the presence of a normal erythropoietin level rules out the diagnosis of PV.3
The presence of a JAK2 V617F in suspected PV is highly supportive of the diagnosis, regardless of the serum erythropoietin level. In the absence of a JAK2 V617F mutation, the serum erythropoietin level is useful to guide further evaluation. If the serum erythropoietin level is subnormal, a JAK2 exon 12 mutation screen should be performed. In the setting of a negative JAK2 mutation and a normal erythropoietin level, the diagnosis of PV is unlikely and evaluation should focus on secondary causes of erythrocytosis.
The oldest PV diagnostic criteria was developed in the late 1960s by the French polycythemia vera study group (PVSG). This was prior to the general availability of assays for erythropoietin and JAK2 mutational analyses, and at a time when blood volume studies were readily available. Today this diagnostic tool has its advantages and limitations. The PVSG diagnostic criteria for PV require the presence of all three major criteria or the presence of the first two major criteria and any two minor criteria:
a. Major criteria
Increased RBC mass: males: ≥36 mL/kg and females: ≥32 mL/kg
Arterial oxygen saturation ≥92%
Splenomegaly
b. Minor criteria
Platelet count >400,000/µL
White blood cell (WBC) count >12,000/µL
Leukocyte alkaline phosphatase score>100
Serum vitamin B12 > 900 pg/mL or serum unbound B12 binding capacity >2,200 pg/mL.
The World Health Organization’s (WHO) revised diagnostic criteria were developed to surpass some of the limitations of the PVSG diagnostic criteria.1 The diagnosis of PV requires meeting either both major criteria and one minor criterion or the first major criterion and two minor criteria:
a. Major criteria
Hemoglobin greater than 18.5 g/dL in men, 16.5 g/dL in women, or other evidence of increased RBC volume
Presence of JAK2 V617F or other functionally similar mutation such as JAK2 exon 12 mutation
b. Minor criteria
BM biopsy showing hypercellularity for age with trilineagemyeloproliferation
Serum erythropoietin level below the normal reference range
Endogenous erythroid colony formation in vitro1
The 2008 WHO diagnostic criteria for PV are currently under revision. Proposed changes in PV include lowering of the diagnostic hemoglobin/hematocrit level to 16.5 g/dL/49% in men and 16 g/dL/48% in women, in the presence of consistent BM morphology, and the inclusion of BM morphology as a major criterion, along with JAK2 mutation screening. Even though WHO revised diagnostic criteria are the most used tool for diagnosing PV, two interesting discussion topics remain unanswered. The first is whether the use of hematocrit rather than hemoglobin as a measuring tool provides a greater predictive value of the RBC volume for diagnosis and evaluation of response to therapy. This position is defended by the clinicians using the revised British Committee for Standards in Hematology’s definition as an alternative definition based on hematocrit. The second diagnostic dilemma pertains to the masked PV phenomena described by Barbui et al., where patients present with normal hemoglobin but with suspicious features as an unexplained thrombosis or itching.5 The important message here for clinicians is to maintain a high degree of clinical vigilance in this subset of patients. Figure 21.1 proposes a potential algorithm to guide clinicians in the diagnosis of PV.
2. Aims of therapy
PV is generally an indolent disorder with the decision to treat on the basis of risk stratification. In patients younger than 60 years, the median survival is approximately 24 years. Risk factors for survival comprise leukocytosis (leukocyte count >13,000/µL [to convert to ×109/L, multiply by 0.001]), thrombosis, advanced age (>70 years), and abnormal karyotype. The respective 10-year relative survival rates for patients with no risk factors, one, and two risk factors were 84%, 59% and 26% respectively.6
In contrast, low-risk patients (i.e., those with no history of thrombosis, age less than 60 years, or platelets below 1 × 106/µL) are usually treated with phlebotomy and/or aspirin (ASA). The goal of phlebotomy is to keep the hematocrit level below 45% in men and below 42% in women. Initially, phlebotomy is used to reduce hyperviscosity by decreasing the RBC mass, and subsequent phlebotomies help maintain the RBC mass in a normal range. For patients with high-risk features (i.e., history of thrombosis, or an age greater than 60 years), treatment consists of phlebotomy, ASA, and cytoreductive therapy. Control of hypertension and diabetes and avoidance of smoking are also important. Although
PV patients generally benefit of good outcomes, the challenges of uncontrolled PV symptoms and refractory disease are imminent clinical problems. To address these challenges a PV management algorithm is proposed in Figure 21.2.
PV patients generally benefit of good outcomes, the challenges of uncontrolled PV symptoms and refractory disease are imminent clinical problems. To address these challenges a PV management algorithm is proposed in Figure 21.2.
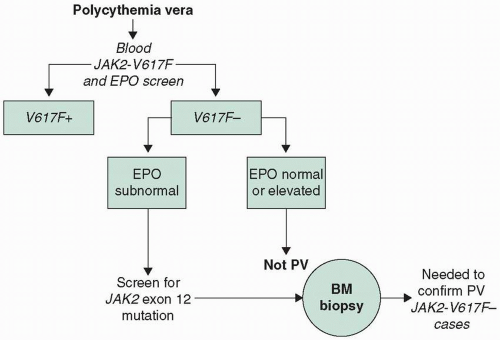 FIGURE 21.1 Proposed diagnostic algorithm for PV. BM, bone marrow; EPO, erythropoietin; PV, polycythemia vera. |
3. Treatment regimens
a. Phlebotomy
Removal of 350 to 500 mL of blood every 2 to 4 days (less often in the elderly or in patients with cardiac disease) is the standard initial approach; the goal is getting the hematocrit to 40% to 45%. The blood count is then checked monthly, and phlebotomy is repeated as needed to maintain the hematocrit at no more than 45%. Rapid lowering of the hematocrit may also be achieved in emergency situations by erythropheresis. Elective surgery should be deferred until the hematocrit has been stable at no more than 45% for 2 to 4 months. Platelet function should be evaluated before surgery or invasive procedures.6
b. Antithrombotic therapy
Concomitantly with phlebotomy, use of low-dose ASA is now widely regarded as standard therapy, following a large European study (European Collaboration on Low-Dose Aspirin in Polycythemia [ECLAP]) utilizing 100 mg ASA daily that showed an approximately 60% reduction in thrombotic
events.7 Higher doses of ASA (325 mg daily) carry risk of bleeding, especially in patients with platelet counts greater than 1.5 × 106/µL, in whom acquired von Willebrand disease may be seen. The exact thrombogenic role of platelets in MPNs is not clear, but hydroxyurea and anagrelide have been shown to lower platelet counts and reduce the risk of thrombosis.6
events.7 Higher doses of ASA (325 mg daily) carry risk of bleeding, especially in patients with platelet counts greater than 1.5 × 106/µL, in whom acquired von Willebrand disease may be seen. The exact thrombogenic role of platelets in MPNs is not clear, but hydroxyurea and anagrelide have been shown to lower platelet counts and reduce the risk of thrombosis.6
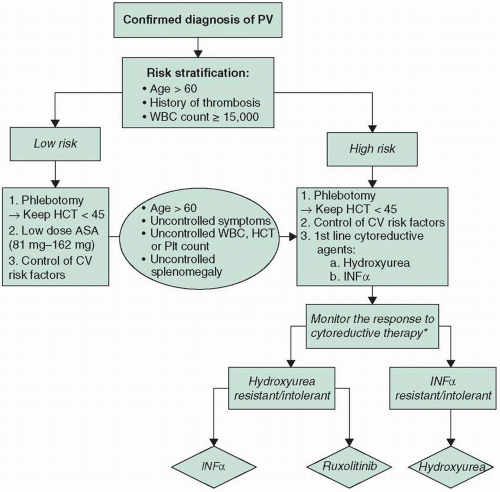 FIGURE 21.2 Management approach for PV. *European Leukemia Net Definition of Resistance/Intolerance to hydroxyurea. ASA, aspirin; CV, cardiovascular; HCT, hematocrit; INFa, interferon α; Plt, platelet; PV, polycythemia vera; WBC, white blood cells. |
c. Myelosuppressive agents
Myelosuppressive agents are indicated in conjunction with phlebotomy for persistent thrombocytosis, recurrent thrombosis, enlarging spleen, or similar problems. They may also reduce the risk of progression to MF compared with phlebotomy alone. Most alkylating agents carry a high risk of inducing a secondary myelodysplastic syndrome (MDS) or
leukemia and should no longer be used.6 Currently recommended choices are as follows:
leukemia and should no longer be used.6 Currently recommended choices are as follows:
1) Hydroxyurea 10 to 30 mg/kg by mouth daily. Weekly blood cell counts are required initially, with dose adjustments to maintain the hematocrit at no more than 45%, the platelet count at 100,000 to 500,000/µL, and the WBC count at greater than 3,000/µL. Side effects are usually minimal, but long-term use may cause painful leg ulcers and aphthous stomatitis. For younger patients and cases difficult to control with hydroxyurea, acceptable alternatives include the following.
2) Interferon-α (IFN-α) is usually effective in controlling hematocrit, platelet count, and splenomegaly and in relieving pruritus. The starting dose is 1 to 3 × 106 U/m2 three times weekly (pegylated interferon once weekly may also be an option—see Section I.C.2.5.). Common side effects include myalgia, fever, and asthenia, usually controlled with acetaminophen. Leukemogenic effects are presumably absent, but high cost is a deterrent to long-term use. For high-risk patients during pregnancy IFN-α is the optimal choice of management. Quintás-Cardama et al. published the results of their phase II trial with pegylated-IFN-α-2a (PEG-IFN-α-2a) treatment in 43 PV patients.8 This study showed that after a median follow-up of 42 months, complete hematologic response (CHR) was achieved in 76% of patients with a median duration to complete response of 40 days. The same study also reported a complete molecular response (CMR) in 18%, partial molecular response (<50% reduction) in 35%, and drug-related treatment discontinuation in 20% of patients. Kiladjian et al. also reported the results of their multicenter phase II trial evaluating the efficacy, molecular response, and safety of PEG-IFN-α-2a in 40 PV patients.9 Their findings demonstrated at 12 months follow-up, 94.6% CHR was sustained for a median follow-up of 31.4 months. Partial molecular response was obtained in 89.6% and CMR in 24.1% of patients, with only 8% of drug-related treatment discontinuation. These two trials demonstrate that PEG-IFN-α-2a induces sustained CHR and CMR with tolerable treatment-related adverse events (AEs) in a subset of PV patients. A multicenter randomized trial is currently comparing the efficacy and safety between hydroxyurea and PEG-IFN-α-2a in high-risk PV patients, which might provide clinicians with the needed evidence to support their choice of first-line agent for highrisk PV patients. On the other hand, a novel investigational mono-pegylated-proline-IFN-α-2b administrated every 14 days subcutaneously (SC) has shown efficacy and safety in a phase I/II trial. The study reported that at 18 months,
there was 47% complete remission (CR) and 42% partial remission, resulting in an overall response rate (ORR) of 89%, and drug-related treatment discontinuation was reported in 20%. The more convenient treatment schedule (once every 14 days) along these results supports further development accordingly, and a phase III clinical study (PROUD-PV) started recruiting in 2013.
there was 47% complete remission (CR) and 42% partial remission, resulting in an overall response rate (ORR) of 89%, and drug-related treatment discontinuation was reported in 20%. The more convenient treatment schedule (once every 14 days) along these results supports further development accordingly, and a phase III clinical study (PROUD-PV) started recruiting in 2013.
3) Radioactive phosphorus (32P) 2.3 mCi/m2 intravenously (IV) (5mCi maximum single dose). Repeat in 12 weeks if the response is inadequate (25% dose escalation optional). Lack of response after three doses mandates a switch to other forms of therapy. Use of 32P entails an approximately 10% risk of leukemia by 10 years, and it is best reserved for the elderly and patients refractory to other modalities. Supplemental phlebotomies may be required for patients with satisfactory platelet and WBC counts but with rising hematocrit levels.
4) Busulfan appears to have less leukemogenic potential than other alkylating agents and is appropriate in patients whose disease is not controlled by other treatments or in the elderly. It is best given in short courses over several weeks (to avoid prolonged marrow suppression) at 2 to 4 mg/day.
5) Anagrelide selectively inhibits platelet production, and platelets start to fall in 7 to 14 days. The WBC count is unaffected; hemoglobin may fall slightly. Responses to anagrelide have been reported in more than 80% of patients with MPNs, and thrombotic risk is reduced. Recommended starting dose is 0.5 mg by mouth once a day. Average dose for control is 2.4 mg daily. Side effects include headache (44%), palpitations, diarrhea, asthenia, and fluid retention. It should be used with caution in cardiac patients and is contraindicated in pregnancy.
6) Ruxolitinib is a potent JAK1/JAK2 inhibitor with a nonspecific myelosuppressive effect and an anti-JAK-STAT-mediated downregulation of inflammatory cytokine activity. Well-documented side effects include, nonexclusively, transaminitis, cholesterol elevation, anemia, and thrombocytopenia. A recent phase III open-label study (RESPONSE trial) evaluated the efficacy and safety of ruxolitinib versus standard therapy in patients with PV who had an inadequate response to, or had unacceptable side effects from, hydroxyurea.10 The study randomly assigned phlebotomy-dependent patients with splenomegaly, in a 1:1 ratio, to receive ruxolitinib or standard therapy. The primary endpoint was both hematocrit control through week 32 and at least a 35% reduction in spleen volume at week 32. The results showed that at week 32, two-thirds of
patients in the ruxolitinib arm were receiving doses of 10 mg twice per day (BID) or 15 mg BID. The primary endpoint was achieved in 21% of the patients in the ruxolitinib group versus 1% of those in the standard-therapy group (p < 0.001).10 Furthermore, hematocrit control was achieved in 60% of patients receiving ruxolitinib and 20% of those receiving standard therapy, while 38% and 1% of patients in the two groups, respectively, had at least a 35% reduction in spleen volume. A complete hematologic remission was achieved in 24% of patients in the ruxolitinib group and 9% of those in the standard-therapy group (p = 0.003). Additionally, 49% of patients taking ruxolitinib had at least a 50% reduction in the total symptom score at week 32, versus 5% of the standard group. Overall, 77% of patients randomized to ruxolitinib met at least one component of the primary endpoint and 91% of patients who met the primary endpoint had a confirmed response at week 48. As for response sustainability, only one patient lost primary response 37.1 weeks after its start with a 94% probability of maintaining primary response for 1 year. On the other hand, the phlebotomy rate at weeks 8 to 32 was more than three times higher in the standard-therapy arm compared with the ruxolitinib arm. Also, only 2.8% of patients required three or more phlebotomies during this time compared with 20.2% in the standard-therapy group. Regarding the exposure-adjusted AEs and grade 3 or 4 AEs per 100 patient-years over the entire course of treatment, they were lower in patients randomized to ruxolitinib compared with those randomized to standard-therapy arm. Exposure-adjusted rates of serious AEs per 100 patient-years were comparable in both arms. In the ruxolitinib group, grade 3 or 4 anemia occurred in 2% of patients, and grade 3 or 4 thrombocytopenia occurred in 5%; the corresponding percentages in the standard-therapy group were 0% and 4%. No patients discontinued treatment because of anemia or thrombocytopenia. Thromboembolic events occurred in one patient receiving ruxolitinib and in six patients receiving standard therapy. This trial demonstrated that ruxolitinib was superior to standard therapy in controlling the hematocrit, reducing the spleen volume, and improving symptoms associated with PV. This study led to the approval of ruxolitinib as the preferred second-line agent in patients resistant or intolerant to hydroxyurea.10,11
patients in the ruxolitinib arm were receiving doses of 10 mg twice per day (BID) or 15 mg BID. The primary endpoint was achieved in 21% of the patients in the ruxolitinib group versus 1% of those in the standard-therapy group (p < 0.001).10 Furthermore, hematocrit control was achieved in 60% of patients receiving ruxolitinib and 20% of those receiving standard therapy, while 38% and 1% of patients in the two groups, respectively, had at least a 35% reduction in spleen volume. A complete hematologic remission was achieved in 24% of patients in the ruxolitinib group and 9% of those in the standard-therapy group (p = 0.003). Additionally, 49% of patients taking ruxolitinib had at least a 50% reduction in the total symptom score at week 32, versus 5% of the standard group. Overall, 77% of patients randomized to ruxolitinib met at least one component of the primary endpoint and 91% of patients who met the primary endpoint had a confirmed response at week 48. As for response sustainability, only one patient lost primary response 37.1 weeks after its start with a 94% probability of maintaining primary response for 1 year. On the other hand, the phlebotomy rate at weeks 8 to 32 was more than three times higher in the standard-therapy arm compared with the ruxolitinib arm. Also, only 2.8% of patients required three or more phlebotomies during this time compared with 20.2% in the standard-therapy group. Regarding the exposure-adjusted AEs and grade 3 or 4 AEs per 100 patient-years over the entire course of treatment, they were lower in patients randomized to ruxolitinib compared with those randomized to standard-therapy arm. Exposure-adjusted rates of serious AEs per 100 patient-years were comparable in both arms. In the ruxolitinib group, grade 3 or 4 anemia occurred in 2% of patients, and grade 3 or 4 thrombocytopenia occurred in 5%; the corresponding percentages in the standard-therapy group were 0% and 4%. No patients discontinued treatment because of anemia or thrombocytopenia. Thromboembolic events occurred in one patient receiving ruxolitinib and in six patients receiving standard therapy. This trial demonstrated that ruxolitinib was superior to standard therapy in controlling the hematocrit, reducing the spleen volume, and improving symptoms associated with PV. This study led to the approval of ruxolitinib as the preferred second-line agent in patients resistant or intolerant to hydroxyurea.10,11
4. Ancillary treatments
To control hyperuricemia, allopurinol 300 mg/day is usually effective. Pruritus is a frequent problem, but usually abates with myelosuppressive therapy. Cyproheptadine 5 to 20 mg/day or
paroxetine 20 mg/day may be helpful; IFN-α is also frequently effective. ASA is often helpful for erythromelalgia (hot, red, painful digits) and is commonly used to prevent thrombosis.7 Use of ASA should be avoided in patients with acquired von Willebrand syndrome, in the presence of less than 20% activity on risotecin cofactor activity assay.
paroxetine 20 mg/day may be helpful; IFN-α is also frequently effective. ASA is often helpful for erythromelalgia (hot, red, painful digits) and is commonly used to prevent thrombosis.7 Use of ASA should be avoided in patients with acquired von Willebrand syndrome, in the presence of less than 20% activity on risotecin cofactor activity assay.
5. Evolution and outcome
The median survival in patients with PV exceeds 15 years, and the 10-year risk of developing either MF (10%) or acute myeloid leukemia (AML; 6%) is relatively low.12 The JAK2 V617F allele burden or JAK2/CALR mutational status has not been proven to affect survival. To date, drug therapy has not been shown to favorably affect these figures. Therefore, at present, drugs should not be used with the intent to either prolong survival or prevent disease transformation into AML or MF. Patients should be preferably monitored by a hematologist, especially when initiated on cytoreductive agents for disease/drug-related side effects and blood count every 2 weeks when disease is active. Later, once the disease is stable, monitoring can be spaced out to every 2 to 3 months. The decrease in WBC and platelet count seems to be most indicative of a response to therapy. Response to therapy criteria have been well established by the European Leukemia Net and a complete response is composed of either (a) hematocrit <45% without phlebotomy or (b) response in three of the following four criteria: (1) platelet count ≤400 × 109/L; (2) WBC count ≤10 × 109/L; (3) normal spleen size on imaging; (4) no disease-related symptoms.
C. Essential thrombocythemia
1. Diagnosis
The median age for diagnosis of ET is 55 to 60 years with a significantly higher incidence in females (female-to-male ratio of up to 2:1). As described previously, around 90% of patients with ET have exclusive mutations in one of three genes that are thought to act as drivers of the disease process. Between 50% and 60% of patients with ET are found to have a mutation in the JAK2 gene, JAK2 V617F, while 5% of patients harbor mutations in the MPL. A more recent advance was the discovery of CALR mutation, found in up to 50% to 70% of patients who are negative for JAK2 or MPL mutations.3 These mutational changes define certain patient characteristics and increased risk of disease-related complications. Indeed, patients with CALR mutation were males, of younger age, and had a lower incidence of thrombosis, hemoglobin level, leukocyte counts, and higher platelet counts, when compared with JAK2 V617F-positive ET. In contrast, patients with the uncommon MPL mutation were found to have lower hemoglobin, higher platelet count, and an increased incidence of thrombosis.
Diagnosis of ET requires a persistent elevation of the platelet count above 450 × 103/µL plus the absence of known causes of reactive or secondary thrombocytosis (e.g., iron deficiency, malignancy, and chronic inflammatory disease). After excluding obvious causes of reactive thrombocytosis (iron deficiency, trauma, infection, and so on), peripheral blood testing for the JAK2 V617F mutation is helpful. The presence of JAK2 V617F confirms clonal thrombocytosis but careful review of the peripheral blood smear and BM histology with cytogenetics must also be performed to confirm the diagnosis of ET.13 Chronic myeloid leukemia (CML) can often present with thrombocytosis; therefore, the presence of BCR-ABL must be excluded. As stated previously, the absence of JAK2 does not rule out the possibility of ET given that a large proportion of patients do not carry the mutation. Only 4% of patients with ET without a JAK2 V617F mutation will have a mutation in the MPL gene; however, if present, it does suggest clonal thrombocytosis. Performing a BM biopsy is mandatory to rule out PMF, MDS/MPN, and “prefibrotic” PMF.13 The BM biopsy in ET will show megakaryocyte proliferation with large and mature morphology. Moderate leukocytosis is common. Palpable splenomegaly is present in less than 50% of patients. Platelet function studies may show either spontaneous aggregation or impaired response to agonists. Microvascular occlusion may cause digital gangrene, transient ischemic attacks, visual complaints, and paresthesias. Large-artery thrombotic episodes are also common. Deep venous thrombosis is uncommon. The risk of hemorrhagic problems is significant, particularly with a platelet count greater than 1,500 × 103/µL.
The diagnosis of ET requires meeting four criteria, as follows:
Sustained platelet count of at least 450 × 103/µL.
BM biopsy specimen showing proliferation mainly of the megakaryocytic lineage with increased numbers of enlarged, mature megakaryocytes. No significant increase or left shift of neutrophil granulopoiesis or erythropoiesis.
Not meeting WHO criteria for PV or PMF, BCR–ABL-positive CML, or MDS or other myeloid neoplasm.
Demonstration of JAK2 V617F or other clonal marker or, in the absence of JAK2 V617F, no evidence of reactive thrombocytosis.1
The 2008 WHO diagnostic criteria for ET are currently being under revision and the proposed changes include the inclusion of CALR mutations as a clonal marker.
2. Treatment regimens
Given its typically indolent course, the primary goal of treatment in patients with ET is the prevention of complications from thrombocytosis, such as microvascular disturbances or
hemorrhagic events caused by acquired von Willebrand disease. ASA therapy is often used to reduce microvascular symptoms for patients with all risk categories. Hydroxyurea, to reduce platelet counts, in combination with low-dose ASA, has been shown to decrease the risk of arterial thrombosis in patients with highrisk ET, such as those older than 60 years with platelets greater than 1,000 × 103/µL or a history of hypertension, diabetes requiring treatment, or ischemia; thrombosis; embolism; or hemorrhage related to ET. For patients with ET whose platelet counts are refractory to therapy with ASA or other salicylates, therapy with IFN-α (including in pegylated preparations), anagrelide, or hydroxyurea can be used. Anagrelide was developed to prevent platelet aggregation but was subsequently found to reduce platelet counts in ET and PMF when used at low dose.
hemorrhagic events caused by acquired von Willebrand disease. ASA therapy is often used to reduce microvascular symptoms for patients with all risk categories. Hydroxyurea, to reduce platelet counts, in combination with low-dose ASA, has been shown to decrease the risk of arterial thrombosis in patients with highrisk ET, such as those older than 60 years with platelets greater than 1,000 × 103/µL or a history of hypertension, diabetes requiring treatment, or ischemia; thrombosis; embolism; or hemorrhage related to ET. For patients with ET whose platelet counts are refractory to therapy with ASA or other salicylates, therapy with IFN-α (including in pegylated preparations), anagrelide, or hydroxyurea can be used. Anagrelide was developed to prevent platelet aggregation but was subsequently found to reduce platelet counts in ET and PMF when used at low dose.
3. Hydroxyurea
A dosage of 10 to 30 mg/kg by mouth daily, with dosage adjustments on the basis of weekly blood counts, should give satisfactory response in 2 to 6 weeks. Its use in combination with low-dose ASA may give optimal protection against arterial thrombosis and evolution to MF.14
4. Anagrelide
Presents a reasonable alternative to hydroxyurea and perhaps is preferable in younger patients. Anagrelide plus ASA was inferior to hydroxyurea plus ASA in a large recent trial.14 This agent should not be used in pregnancy.
5. IFN-α
Along with busulfan, it is considered to be a second-line drug of choice in patients who are resistant to or intolerant of hydroxyurea. Both controlled and uncontrolled single-arm studies have demonstrated long-term safety and efficacy of these drugs. Most patients with ET respond to this agent, at an initial dose of 3 × 106 U/day SC. Maintenance doses of 3 × 106 U three times weekly usually suffice. Pegylated interferon at an initial dose of 1.5 to 4.5 µg/kg/week SC seems comparable in efficacy and side effects. Use of interferon in pregnancy is considered safe.
6. 32P and alkylating agents
These agents are effective but carry increased risk of secondary leukemia. Nitrogen mustard (mechlorethamine 0.15 to 0.3 mg/kg [6 to 12 mg/m2] IV) can be helpful when rapid reduction in platelet count is needed. Busulfan 2 to 4 mg/day initial dose is appropriate in selected elderly patients resistant to other agents.
7. ASA
A dosage of 81 to 325 mg/day may control erythromelalgia and similar vaso-occlusive problems but is contraindicated in patients with a history of hemorrhagic symptoms. ASA may be useful in pregnant patients, in whom the preceding agents are contraindicated. Twice-daily ASA 81 mg use in low-risk patients
whose microvascular symptoms are resistant has been proven to be more efficacious than once-daily ASA. In patients with severe thrombocytosis, ASA should be avoided because of the increased risk of bleeding secondary to acquired von Willebrand disease.
whose microvascular symptoms are resistant has been proven to be more efficacious than once-daily ASA. In patients with severe thrombocytosis, ASA should be avoided because of the increased risk of bleeding secondary to acquired von Willebrand disease.
8. Evolution and outcome
The course of ET is often indolent, particularly in young patients. The median survival is 24 years for patients older than 60 years, and 33 years for those younger than 60. The JAK2/CALR mutational status or JAK2 V617F allele burden has not been shown to affect survival in ET. The only factors predictive of complication development are age >60 and previous thrombotic events. Interestingly, higher platelet levels did not correlate with increased risk of thrombotic events. The international prognostic score of thrombosis in essential thrombocythemia (IPSET-thrombosis) was proposed to better assess the risk of thrombosis. The final prognostic score included four risk factors: age ≥60 years (1 point), cardiovascular risk factors, such as diabetes, hypertension, or smoking (1 point), prior thrombosis (2 points), and JAK2 V617F mutation (2 points). Patients could be stratified into three risk categories as follows: low risk (0 to 1 point), intermediate risk (2 points), and high risk (≥3 points). The annual rate of thrombosis ranged from 1.03% of patients/year in the low-risk category to 3.56% of patients/year in the high-risk category. Another prognostic score was recently developed to predict overall survival (OS) at diagnosis (IPSET) including age ≥60 years (2 points), leukocyte count ≥11 × 109/L (1 point), and prior thrombosis (1 point) as independent risk factors for survival. Patients could again be stratified into three risk categories, with median survival not yet reached in the low-risk group and 14.7 years for the high-risk group. Transformation to PMF, MDS, or acute leukemia occurs in 5% to 10% of patients with ET. Thrombosis remains the major cause of ET-related death.15
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


