Myelodysplastic syndromes (MDS) are spectrum of bone marrow failure disorders that share a common pathologic feature: cytologic dysplasia. The classification of MDS reflects the understanding of the disease. It is hoped that in the future classification and risk stratification will be based on underlying pathobiology of different disease subsets and molecular signatures where the pathologic classification represents their phenotype. This article reviews MDS classification and risk stratification highlighting differences between the various systems.
Myelodysplastic syndromes (MDS) are bone marrow failure disorders with resultant cytopenias and a tendency to progress to acute myeloid leukemia (AML). The diagnosis of the disease requires demonstration of cytologic dysplasia in one or more of the different bone marrow cell lines.
MDS is more than one disease or subsets of disorders with different driving biologic features that share a common pathologic phenotype “dysplasia.” The classification of MDS directly reflects the understanding of the disease and it has been an evolving process over many years.
At the beginning of the past century the observation was made that a group of patients had a form of anemia refractory to available treatments at that time. The term “anemia pseudoaplastica” was one of the earliest descriptions for MDS. In the late 1940s, it was thought that the disease progresses to leukemia and the term “preleukemia” was applied. The definition was expanded to include multilineage cytopenia in the 1950s. It not until the 1970s when Saarni and Linman recognized the disease as a primary bone marrow disorder characterized by decreased hematopoietic precursor maturation and bone marrow hypercellularity. The French-American-British group (FAB) coined the term “myelodysplastic syndromes” in a series of proposals on the acute leukemias in 1976 and later expanded the FAB classification in the 1980s. The next advancement in classification and risk stratification was in 1997 with the introduction of the International Prognostic Scoring System (IPSS). Under the auspice of the World Health Organization (WHO) a distinguished panel of more than 100 hematologists and hematopathologists revised the MDS classification and published the WHO MDS classification in 1999. In the last decade the evolution of the classification progressed more rapidly with better technologies to understand the disease biology. New prognostic models were introduced, such as the WHO Prognostic Scoring System (WPSS), and the WHO classification was refined.
In this modern era, risk stratification based more on biology is crucial. A better classification will allow the tailoring of treatments toward more homogenous groups of patients. Newer therapies may help in the exploration of underlying disease biology and enhance classification.
Diagnostic criteria for MDS
The general recommendations for diagnosis of MDS include demonstration of cytologic dysplasia in one or more cell lines in patients with persistent or progressive cytopenia or demonstration of increased myeloblasts (5%–19%) ( Fig. 1 ). MDS remains a diagnosis of exclusion. In certain cases MDS diagnosis can be made without clear evidence of morphologic dysplasia, such as chronic myelomonocytic leukemia (CMML) if monocytes are persistently elevated or if a recurrent cytogenetic abnormality is detected in patients with sustained unexplained cytopenia (“presumptive MDS” by WHO 2008 criteria).
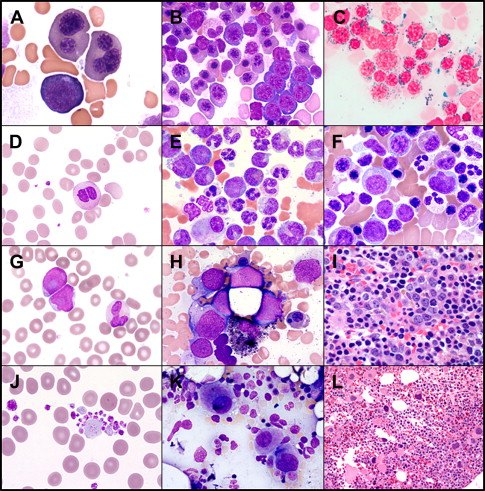
It is still recommended to perform a 500-cell differential, eliminating lymphocytes and plasma cells for the count. The dysplasia in each cell line is noted and the percentage of blasts is calculated. If the absolute percentage of erythroid precursors is 50% or greater, the percentage of blasts is based on the nonerythroid precursors. Iron stains are essential to address the percentage of ring sideroblasts. The bone marrow core biopsy could complement the bone marrow aspirate in several aspects, such as cellularity; dysmegakaryopoiesis; and identifying clusters of abnormal localization of immature precursors, which are immature cells myeloblasts or promyelocytes (displaced from the paratrabecular area to the intertrabecular areas). In addition, the reticulin stain provides additional information about the degree of reticulin fibrosis, which can add prognostic information. Conventional cytogenetics obtained from bone marrow aspirate could be complemented by interphase fluorescence in situ hybridization tests that could be obtained from bone marrow or peripheral blood (PB) samples.
MDS pathologic classifications
FAB Classification
The FAB classification was the first attempt at systemic classification of the disease based on observations and understanding of the disease at that time. It was known that some patients present with a disease that bore some resemblance to AML, but did not have as many myeloblasts in the bone marrow. The disease resulted in alteration in maturation of the three major cell lines, manifesting as pancytopenia. It was also observed that the rate of disease progression was highly variable: some patients never evolved to acute leukemia, whereas others unfortunately did quickly. The original FAB classification included two of the subtypes of what are now called MDS refractory anemia with excess blasts (RAEB) and CMML.
In 1982, a revision of the FAB classification was published. This was based on the accumulating experience with larger number of cases reviewed and observations of different subsets, which have dysplasia as a common feature but had a distinct clinical outcome. This revision included five subgroups: (1) refractory anemia (RA), (2) refractory anemia with ring sideroblasts (RARS), (3) RAEB, (4) RAEB in transformation (RAEB-t), and (5) CMML ( Table 1 ).
| FAB | WHO | WHO 2008 | Dysplasia | BM Blasts % | PB Blasts % |
|---|---|---|---|---|---|
| RA | RA MDS-U RCMD Del5q MDS | RCUD RA RN, RT RCMD Isolated del 5q MDS-U | Erythroid Nonerythroid Erythroid + other Erythroid + mega Unilineage + pancytopneia or RCMD/RCUD with 1% PB blasts | <5 <5 <5 <5 <5 | <1 <1 <1 <1 1 |
| RARS | RARS RCMD-RS | RARS RCMD-RS | Erythroid only Erythroid + other (all >15% ring sideroblasts) | <5 <5 | < 1 < 1 |
| RAEB | RAEB-1 RAEB-2 | RAEB-1 RAEB-2 | ≥1 lineage ≥1 lineage | 5–9 10–19 | 2–4 5–19 Auer rods |
| RAEB-t | AML | AML | Myeloid ± other | ≥20 | |
| CMML | MDS/MPD CMML JMML aCML MDS/MPD-U | MDS/MPN CMML JMML BCR/Abl neg CML MDS/MPD-U | Variable >1 × 10 9 /L monocytosis | <20 |
The RA included any patient with anemia or variant cytopenias with less than 5% myeloblasts and less than 15% ring sideroblasts. The RARS group included patients who demonstrated 15% or more ring sideroblasts (iron from the Greek) where iron stains in necklace fashion around the nucleus. RARS case reports were originally described in 1956. It was later noted that some patients had pure sideroblastic anemia, whereas others had dysplasia in other cell lines and in general had higher risk to transform to AML. RAEB included patients with 5% to 20% myeloblasts in the bone marrow or 1% to 4% blasts on PB, whereas RAEB-t described patients with 20% to 30% myeloblasts in the bone marrow and greater than or equal to 5% blasts on PB or with presence of Auer rods. Finally, CMML was defined by persistent absolute monocytosis on PB (more than 1 × 10 9 /L). The FAB group suggested distinguishing two CMML subtypes: myelodysplastic CMML (white blood cell [WBC] count <13 × 10 9 /L) and myeloproliferative CMML (WBC count ≥13 × 10 9 /L).
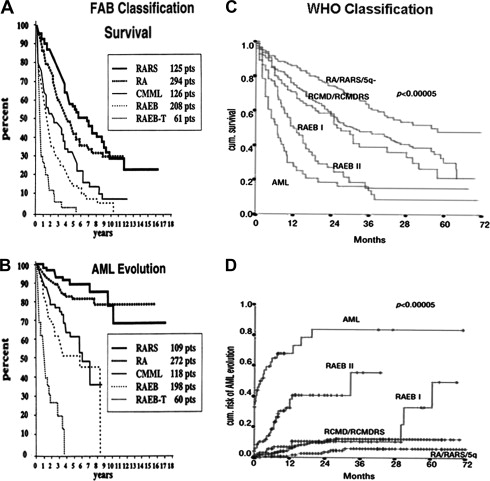
The FAB classification could clearly differentiate outcome between the low-risk group (RA, RARS) and high-risk group (RAEB, RAEB-t) with median overall survival ranging from 5 to 6 years for low risk compared with less than 1 year for the least favorable forms ( Fig. 2 A, B). The classification, however, had several limitations. It did not address the importance of unilineage versus multilineage dysplasia, it did not provide any role for cytogenetic classification, RAEB included a large heterogeneous category and RAEB-t represented a gray area between MDS and AML, and CMML behavior and outcome were not well characterized.
WHO Classification
The WHO classification was an attempt to address some of the shortcomings of the FAB classification creating more homogenous subtypes. The following are the highlights of WHO classification (see Table 1 ).
Recognition of unilineage versus multilineage dysplasia
RA and RARS were divided based on line of dysplasia. Pure RA or RARS was redefined when dysplasia is truly limited to erythroid cell line. The separate categories refractory cytopenia with multilineage dysplasia and refractory cytopenia with multilineage dysplasia and ring sideroblasts (RCMD-RS) were added when more than 10% dysplastic changes are seen in either myeloid or megakaryocytic cell line in addition to the erythroid dysplasia.
This was based on logical observations where presence of multilineage dysplasia carries a worse prognosis even in presence of sideroblasts. A higher frequency and different cytogenetic complexity were reported in patients with RAEB and RCMD compared with RA and RARS. The presence of cytogenetic abnormalities was noted in 39.3% in RA and RARS compared with 50.4% in RCMD patients. Some studies showed the presence of clonal cell subpopulations in the PB and bone marrow of RCMD but not in RA and RARS or del(5q) patients. The WHO recognized cases with unilineage or bilineage myeloid or megakaryocytic dysplasia without erythroid dysplasia but they were categorized as MDS unclassified.
Introduction of cytogenetic information as first attempt of better biologic classification
The WHO classification recognized 5q syndrome as a distinct subgroup with isolated del (5q) and less than 5% myeloblasts. Clinically, this syndrome is characterized by macrocytic anemia and normal to elevated platelets count. It carries a favorable prognosis and it is uncommon that it progresses to AML.
Refining the arbitrary cutoff for blasts percentage to better predict prognosis
The RAEB was divided into RAEB I with 5% to 9% bone marrow blasts and RAEB II with 10% to 19% blasts. The RAEB-t is omitted and the threshold for diagnosing AML was lowered to 20% myeloblasts instead of 30%. The presence of Auer rods was disregarded. The rational for lowering the threshold for AML was that patients with RAEB-t had similar response to treatment as AML, although acknowledging that progression to AML is not a must in each single case. It is very important to note that this classification is prognostic but does not dictate the treatment for this group; most young patients who meet this cutoff are treated as AML but many elderly patients are still treated as MDS.
Recognition of a group of disorders that share dysplasia in common but also may have proliferative behavior
This newly proposed group of diseases was named myelodysplastic-myeloproliferative disorders. It included chronic CMML, juvenile myelomonocytic leukemia, and atypical chronic myelogenous leukemia. The CMML is further classified based the percentage of bone marrow blasts rather than WBC count, where CMML type 1 included 0% to 4% blasts or promonocytes on PB or less than 10% in bone marrow and type 2 included 5% to 19% blasts or promonocytes on PB or less than 20% in bone marrow.
Several studies confirmed the prognostic value of WHO, whereas only a few did not. The first study was a retrospective validation using the Dusseldorf MDS registry. Later, the same group prospectively validated the classification ( Fig. 2 C, D). Among 1095 patients, the median overall survival for RA and RARS was not reached, median overall survival for RCMD was 31 months, RCMD-RS was 28 months, RAEB-I was 27 months, RAEB- II was 12 month, and del (5q) was 40 months. The frequency of AML progression 2 years after diagnosis was RA, 0%; RARS, 8%; RCMD, 9%; RCMD-RS, 12%; RAEB- I, 13%; RAEB- II, 40%; and 5q, 8%.
The WHO introduction of multilineage dysplasia may also have therapeutic implications. Differences in response rates to erythropoietin-stimulating agents and growth factors among different WHO subtypes were reported. The response rate was 67% in RA versus 50% in RCMD. More difference was observed between RARS (75%) and RCMD-RS (9%) ( P value 0.003).
Revised WHO Classification (2008)
For the first time, the WHO 2008 classification proposes diagnosis of “presumptive MDS” in cases with persistent clinical cytopenias without dysplasia if certain cytogenetic abnormalities are present. The WHO categorizes this entity under MDS-U. Although not part of the WHO classification, the term “idiopathic cytopenia of unknown significance” is applied if patients had persistent cytopenias without alternative explanation, no dysplasia, and without the specific cytogenetic abnormalities. Box 1 summarizes the revisions under WHO 2008 classification (see Table 1 ).
- 1.
The revision recognizes less common cases of MDS with unilineage nonerythroid dysplasia. Instead of RA, the term “refractory cytopenia with unilineage dysplasia” (RCUD) is introduced. This category includes RA, refractory neutropenia (RN), and refractory thromobocytopenia (RT). RCUD includes cases where dysplasia is demonstrated in ≥10% of one cell line. The patients have unicytopenia or bicytopenia but not pancytopenia.
- 2.
Patients with pancytopenia and unilineage dysplasia, patients with no overt dysplasia but cytogenetic evidence of MDS, and cases of RCUD or RCMD with PB blasts of 1% and bone marrow blasts less than 5% and peripheral blasts are subsets of cases still categorized as MDS-U.
- 3.
The term “5q syndrome” is less emphasized in the new WHO classification given that lenalidomide therapeutic benefit is the same for patients with 5q syndrome or isolated del 5(q).
- 4.
The revised classification further refines the blasts percent cutoff in higher-risk disease. RAEB-1 now includes cases with 2%–4% PB blasts and RAEB-II includes 5%–19% PB blasts. Auer rods presence regardless of blast percentage qualifies RAEB-II or CMML-2 classification.
- 5.
Most cases of MDS with myelofibrosis (MDS-F) are RAEB cases. Accurate estimate of blasts percentage is difficult and fibrosis is associated with worse outcome.
- 6.
The MDS-MPN category is also refined. The name is changed to myelodysplastic syndromes–myeloproliferative neoplasm (MDS-MPN). Provisional entity of RARS-T (refractory anemia with ring sideroblasts with thrombocytosis) is proposed. Up to 60% of patients harbor the JAK2 V617F mutation. The platelet threshold is 450,000/mm 3 and more. This group of patients tends to have indolent course similar to RARS. Atypical CML is renamed as atypical CML, BCR/ABL-1 negative to emphasize importance of obtaining those tests. A subset of patients with CMML and eosinophilia who are associated with genetic abnormalities including PDGFR are better classified as myeloid neoplasms with eosinophilia. Those patients can respond to imaitinb mesylate therapy.
- 7.
Provisional entity of refractory cytopenia of childhood is proposed.
MDS pathologic classifications
FAB Classification
The FAB classification was the first attempt at systemic classification of the disease based on observations and understanding of the disease at that time. It was known that some patients present with a disease that bore some resemblance to AML, but did not have as many myeloblasts in the bone marrow. The disease resulted in alteration in maturation of the three major cell lines, manifesting as pancytopenia. It was also observed that the rate of disease progression was highly variable: some patients never evolved to acute leukemia, whereas others unfortunately did quickly. The original FAB classification included two of the subtypes of what are now called MDS refractory anemia with excess blasts (RAEB) and CMML.
In 1982, a revision of the FAB classification was published. This was based on the accumulating experience with larger number of cases reviewed and observations of different subsets, which have dysplasia as a common feature but had a distinct clinical outcome. This revision included five subgroups: (1) refractory anemia (RA), (2) refractory anemia with ring sideroblasts (RARS), (3) RAEB, (4) RAEB in transformation (RAEB-t), and (5) CMML ( Table 1 ).
| FAB | WHO | WHO 2008 | Dysplasia | BM Blasts % | PB Blasts % |
|---|---|---|---|---|---|
| RA | RA MDS-U RCMD Del5q MDS | RCUD RA RN, RT RCMD Isolated del 5q MDS-U | Erythroid Nonerythroid Erythroid + other Erythroid + mega Unilineage + pancytopneia or RCMD/RCUD with 1% PB blasts | <5 <5 <5 <5 <5 | <1 <1 <1 <1 1 |
| RARS | RARS RCMD-RS | RARS RCMD-RS | Erythroid only Erythroid + other (all >15% ring sideroblasts) | <5 <5 | < 1 < 1 |
| RAEB | RAEB-1 RAEB-2 | RAEB-1 RAEB-2 | ≥1 lineage ≥1 lineage | 5–9 10–19 | 2–4 5–19 Auer rods |
| RAEB-t | AML | AML | Myeloid ± other | ≥20 | |
| CMML | MDS/MPD CMML JMML aCML MDS/MPD-U | MDS/MPN CMML JMML BCR/Abl neg CML MDS/MPD-U | Variable >1 × 10 9 /L monocytosis | <20 |
Related posts:
 The Epidemiology of Myelodysplastic Syndromes
The Epidemiology of Myelodysplastic Syndromes
 Immunosuppression for Myelodysplastic Syndrome: How Bench to Bedside to Bench Research Led to Success
Mouse Models of Myelodysplastic Syndromes
Hematopoietic Stem Cell Transplantation for MDS
Immunosuppression for Myelodysplastic Syndrome: How Bench to Bedside to Bench Research Led to Success
Mouse Models of Myelodysplastic Syndromes
Hematopoietic Stem Cell Transplantation for MDS
 Lenalidomide for Treatment of Myelodysplastic Syndromes: Current Status and Future Directions
Lenalidomide for Treatment of Myelodysplastic Syndromes: Current Status and Future Directions
 Patient Selection for Transplantation in the Myelodysplastic Syndromes
Patient Selection for Transplantation in the Myelodysplastic Syndromes
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


