62 Mycosis Fungoides
Epidemiology, Etiology, Genetics, and Cytogenetic Abnormalities
Mycosis fungoides is a rare lymphoma. The estimated annual incidence rate in the United States is only approximately 0.64 per 100,000, or fewer than 1000 new cases diagnosed each year.1 It accounts for only 2% of new cases of non-Hodgkin lymphoma. Although it is uncommon, it is the most common of the primary cutaneous T-cell lymphomas (CTCLs) and is distinguished from other CTCLs by its unique clinical and histologic features. It commonly affects older adults (median age 55 to 60 years); however, it may present in younger individuals with similar clinical findings and course.2 There is a 2 : 1 male predominance, without an established racial predilection.
The causal factors of mycosis fungoides and Sézary syndrome are unknown. Some retrospective studies have suggested an causal role for environmental chemical exposure, as a source of either chronic antigenic stimulation or toxic exposure. However, case-controlled studies failed to reveal any relation between occupational or recreational exposures to chemicals and the development of mycosis fungoides, refuting this hypothesis.3,4
A viral cause for mycosis fungoides was once proposed, based on the isolation of human T-cell leukemia/lymphoma virus 1 (HTLV-1) from the peripheral blood lymphocytes of a patient with a cutaneous lymphoma that resembled mycosis fungoides.5 However, it was later demonstrated that this patient actually had a peripheral T-cell lymphoma with skin involvement. The clinical characteristics of this distinct entity, HTLV-1–associated T-cell lymphoma, have now been described more precisely and are quite different from those of typical mycosis fungoides.
A few studies have demonstrated histocompatibility antigen associations linked to mycosis fungoides and Sézary syndrome, in particular Aw31, Aw32, B8, Bw38, and DR5.6,7 The significance of these immunogenetic findings is unclear, but may suggest a genetic predisposition. Specific chromosomal abnormalities also have been demonstrated in some cases, mostly deletions and translocations in chromosome 1 or 6. In one series, the presence of clonal abnormalities in 11 of 19 patients correlated with advanced-stage disease and a significantly reduced survival.8 In a second study, the detection of a chromosomal clone preceded relapse or progression of the disease.9
Cytokines have been implicated in the pathophysiology of mycosis fungoides and Sézary syndrome.10–12 However, whether cytokine abnormalities are primarily involved or are secondary processes in the pathogenesis is unclear. Studies have reported that soluble interleukin-2 (IL-2) receptor (sIL-2R) values in Sézary syndrome were significantly higher than for other malignant or inflammatory T-cell diseases and that the serum levels correlated with clinical course and Sézary cell count. The highest sIL-2R levels were found in patients with advanced disease.12 Other investigators have shown that peripheral blood mononuclear cells from patients with the Sézary syndrome expressed higher levels of IL-4 and lower levels of IL-2 and interferon (IFN) gamma after phytohemagglutinin stimulation, compared with normal controls.10
A number of studies have suggested that the malignant T cells in Sézary syndrome account for aberrant cytokine production, with increased production of T helper type 2 cytokines (e.g., IL-4, IL-5, IL-10) and decreased production of T helper type 1 cytokines (e.g., IL-2 and IFN gamma).13,14 Moreover, this aberrant cytokine production may cause the immune abnormalities seen occasionally in Sézary syndrome. These immune abnormalities may include decreased T-cell responses to antigens and mitogens, impaired cell-mediated cytotoxicity, including natural killer cell and lymphokine-activated killer cell activities, increased levels of serum immunoglobulin E (IgE) and IgA, and peripheral eosinophilia.
Pathologic Conditions
Skin biopsy with routine histologic examination is still considered the most important study to assist the clinician in establishing the diagnosis. The characteristic histopathologic findings of mycosis fungoides demonstrate abnormal cells infiltrating the epidermis (epidermotropism) as single cells or in clusters (Pautrier microabscesses) (Fig. 62-1). Typically, there is also an upper dermal infiltrate that includes cells similar to those seen in the epidermis, as well as variable proportions of histiocytes, eosinophils, and plasma cells. The criteria for a diagnosis of mycosis fungoides vary among pathologists. Based on the severity of the epidermal and dermal involvement, the categories “diagnostic of,” “consistent with,” and “suggestive of” mycosis fungoides have been recommended.15 Treatment programs specific for mycosis fungoides should be considered only in patients who have a biopsy that is diagnostic of or consistent with, not merely suggestive of, mycosis fungoides.
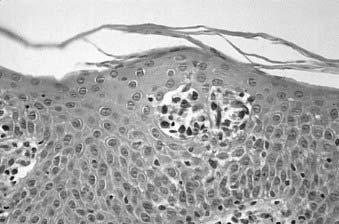
Immunoperoxidase staining indicates that the majority of cases of mycosis fungoides are associated with the helper T-cell phenotype (CD4+) and are also positive for CD2, CD3, CD45, and CD5. These cells may also be positive for CD25 and the p55 (alpha chain) subunit of the IL-2 receptor.16 Usually, although the neoplastic cells of mycosis fungoides retain the CD4 antigen on their cell surface, they lose other mature T-cell antigens, such as CD26 or Leu-9 (CD7). The loss of these mature T-cell antigens may help in the differential diagnosis of mycosis fungoides from benign dermatoses. Rare cases of mycosis fungoides have been demonstrated to be CD8+ (cytotoxic/suppressor T-cell phenotype).
Evaluation of skin biopsies to detect T-cell receptor (TCR) gene rearrangements (genotyping) can be helpful in the differential diagnosis of early mycosis fungoides. TCR gene rearrangements can be detected by Southern blot analysis17 or by methods using polymerase chain reaction (PCR) amplification.18 PCR amplification methods for frozen or paraffin tissue analysis are widely available and affordable. Genotyping is becoming an important diagnostic procedure whenever the routine histologic examination or immunophenotyping is equivocal in patients whose clinical presentation is strongly suggestive of mycosis fungoides or Sézary syndrome.
The pathologic characteristics of extracutaneous disease pose special problems. In the most common situation, enlarged lymph nodes may be biopsied and demonstrate changes of dermatopathic lymphadenitis, including the presence of sinus histiocytosis and an abundance of pigment-laden macrophages. In addition, there may be a variable number of atypical lymphocytes with cerebriform nuclei. The prognostic relevance of different degrees of infiltration by these abnormal cells led to the development of a lymph node classification system. In this system, lymph nodes are classified as LN0 to LN4. Category I (LN0–2) includes dermatopathic nodes and nodes with clusters of less than six atypical cells, category II (LN3) designates lymph nodes with clusters of 10 or more atypical cells, and category III (LN4) includes nodes that are partially or completely effaced by atypical cells.19 Detection of abnormal cells in the lymph nodes is facilitated by the use of Southern blot or PCR analysis. Potential neoplastic involvement with clonal TCR rearrangement may be demonstrated even in lymph nodes that show only dermatopathic changes on routine evaluation.19,20
Flow cytometry studies of the peripheral blood may show expansion of the CD4+/CD7− population reflective of circulating atypical lymphocytes of Sézary type.21 Patients may have atypical lymphocytes with cerebriform nuclei, the Sézary cells, in their peripheral blood. It is controversial as to how many or what percentage of Sézary cells constitutes a significant level to define the Sézary syndrome, and low levels of Sézary-like cells can be detected in the peripheral blood of patients with benign skin conditions.22 Although the original National Cancer Institute (NCI) classification used the criterion of greater than 5% of lymphocytes for significant blood involvement,23 the current practice by many mycosis fungoides referral centers is to use an absolute Sézary cell count of more than 1000/μL or a CD4+/CD8+ T-cell ratio of more than 10 for defining peripheral blood involvement.
Clinical Presentation
Mycosis fungoides often has a long natural history, and the median duration from the onset of skin symptoms to a diagnosis of mycosis fungoides may be 5 years or longer.24 In many patients, the disease presents initially in a premycotic phase with nonspecific, slightly scaling skin lesions that wax and wane for years. Biopsies are generally nondiagnostic during this phase of disease, and patients may respond to treatment with topical corticosteroids. Some of these patients experience an evolution of their disease and develop more typical patches or infiltrated plaques, from which a definitive diagnostic biopsy may be obtained. Repeated biopsies must be obtained from patients suspected of having mycosis fungoides, even when an initial biopsy is negative.
Infiltrated plaques may evolve into ulcerating or fungating tumors; however, the rapidity of this progression is unpredictable. The majority of patients with initial patch or plaque disease who have been treated at Stanford University have never progressed to have more advanced cutaneous disease.25,26 Cutaneous tumors may become infected, and sepsis may be the cause of death in individuals so affected. Occasional patients present de novo with tumors, so-called tumor d’emblée. Generalized dermal thickening from infiltrative disease may cause the classic but very unusual leonine facies of mycosis fungoides.
Another manifestation of skin involvement in mycosis fungoides is generalized erythroderma. The erythema may be accompanied by either atrophic or lichenified skin, and plaques or tumors may also be present. These patients are almost always intensely symptomatic from pruritus and scaling, and often have lymphadenopathy caused by diffuse and severe skin involvement. Many of these patients also have circulating abnormal cells in the peripheral blood that have the same microscopic appearance, immunophenotyping, and genotyping characteristics as the cells that infiltrate the epidermis. Patients with this complex of findings, generalized erythroderma, lymphadenopathy, and atypical T cells (Sézary cells) in the peripheral blood have Sézary syndrome.27 Patients with Sézary syndrome have a worse prognosis than erythrodermic patients with mycosis fungoides who do not have the other findings of the Sézary syndrome.
Routes of Spread
Many patients with mycosis fungoides have evidence of cutaneous disease only throughout their lifetime. Although molecular studies may reveal extracutaneous disease to be present in a significant proportion of patients, only 15% to 20% of patients with mycosis fungoides develop clinical problems related to extracutaneous disease. The likelihood of developing extracutaneous disease is related to the extent of skin involvement. In a series of patients at Stanford with extracutaneous disease, at that time none had limited plaque disease, 11 had generalized plaque disease, 39 had cutaneous tumors, and 27 had erythroderma.28 The most commonly identified route of extracutaneous spread of mycosis fungoides is to the regional lymphatics, usually in areas that drain significant sites of skin involvement. Visceral disease may be identified subsequently. The most common visceral sites of involvement identified are the lungs, the oral cavity and pharynx, and the central nervous system, but virtually any organ may be involved at autopsy in patients who have died of the disease.29
The risk of disease progression in 525 patients with mycosis fungoides treated at Stanford has been analyzed. Patients were considered to have had disease progression when one of the following events occurred: progression of their mycosis fungoides to a more advanced tumor-node-metastases-blood (TNMB) classification, a more advanced clinical stage, or death resulting from mycosis fungoides. The actuarial risk of disease progression at 10 years was 13% in limited plaque, 32% in generalized plaque, 72% in tumorous, and 57% for patients with erythroderma.30
The actuarial risk for developing extracutaneous disease in our 491 patients who presented with stage I to III disease was analyzed according to their initial T classification. The risk at 10 years was 2% in limited plaque, 9% in generalized plaque, 40% in tumorous, and 10% in patients with erythroderma. However, at the time these patients developed extracutaneous disease, none had limited plaque disease.30
Diagnostic and Staging Studies
The standard staging evaluation for patients with mycosis fungoides includes a comprehensive physical examination with careful examination of the skin (including the scalp, palms, soles, and perineum) and lymph nodes, a complete blood count with Sézary cell studies, screening chemistries (including lactate dehydrogenase), and chest x-ray examination. Based on observations that it is unlikely for patients with T1 or T2 skin involvement to present with extracutaneous disease at diagnosis, additional imaging studies (computed tomography [CT] or magnetic resonance imaging) or nuclear medicine scans (positron emission tomography [PET]) are not recommended unless the patient has clinically significant lymphadenopathy. Because patients with T3 or T4 disease are at greater risk for extracutaneous involvement, further imaging studies should be considered. At Stanford we obtain a CT scan of the chest, abdomen, and pelvis or total-body integrated PET-CT scan in these patients.31 Lymph node biopsies should be obtained if lymphadenopathy is present, because the presence of lymph node involvement affects the stage, prognosis, and management. Suspected sites of visceral involvement must be confirmed by appropriate imaging studies and biopsy when possible. Bone marrow involvement may often be detected in patients who meet the clinical criteria for Sézary syndrome,32 but is extremely uncommon in classical mycosis fungoides. Therefore, a bone marrow biopsy is not routinely used as part of the initial staging procedure for patients with mycosis fungoides.
Staging System
A TNMB staging system that has proved useful for mycosis fungoides was proposed at the Workshop on Mycosis Fungoides held at the NCI in 1978.23 This staging system has been revised to reflect updated prognostic information and be more consistent with current practice.33 Table 62-1 and Table 62-2 summarize the classification and TNMB categories.
Table 62-1 TNMB Classification for Mycosis Fungoides (AJCC 2010)
Rights were not granted to include this table in electronic media. Please refer to the printed book.

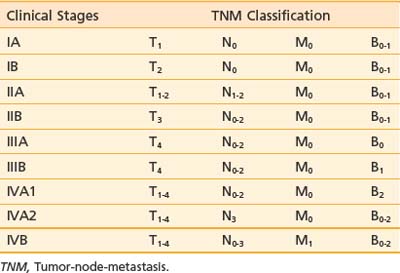
The T classification reflects the extent of skin involvement. The N classification indicates the presence of lymph node involvement. Enlarged lymph nodes should be biopsied (excisional biopsies are recommended), because palpable enlargement is often associated only with changes of dermatopathic lymphadenitis; however, patients with lymph nodes exhibiting rearranged TCR genes have a worse prognosis, regardless of the histologic grade.33 In the M classification, suspected disease should also be documented and treatment programs for visceral disease should be considered only if there is definite proof of extracutaneous disease. In the B classification, the presence of a significant proportion of abnormal, cerebriform (Sézary) cells should be noted; however, low levels of Sézary-like cells can be detected in the peripheral blood of patients with benign skin conditions. The current practice is to use the criterion of an expanded peripheral blood CD4+ population with increased ratio of CD4/CD8 T lymphocytes (greater than 10:1), expanded populations of abnormal T cells with CD4+/CD7− or CD4+/CD26− phenotype, and molecular evidence of a relevant TCR gene rearrangement in the peripheral blood.33
Standard Therapeutic Approaches
Multiple therapeutic options exist for mycosis fungoides and the Sézary syndrome. The National Comprehensive Cancer Network (NCCN) has established consensus guidelines for the therapy of these diseases (www.nccn.org, non-Hodgkin lymphoma mycosis fungoids/Sézary syndrome practice guidelines). Selection of treatment is based primarily on the clinical stage of the disease (Fig. 62-2). However, factors such as access to special treatment approaches, the patient’s age, other social and medical problems, and the cost/benefit ratio should also be taken into consideration. For patients with patch or plaque skin involvement (T1 and T2) without extracutaneous disease, the treatment plan may be limited to topical therapeutic measures, whereas patients with any extracutaneous disease should receive systemic (cytotoxic or biologic) therapy as part of their management. There is no evidence that early aggressive combined modality therapy is more effective than conservative sequential therapies in the management of limited or advanced disease.34 Despite decades of experience in the treatment of mycosis fungoides and Sézary syndrome, well-designed, prospective, controlled clinical studies comparing the efficacy of various therapies are lacking.
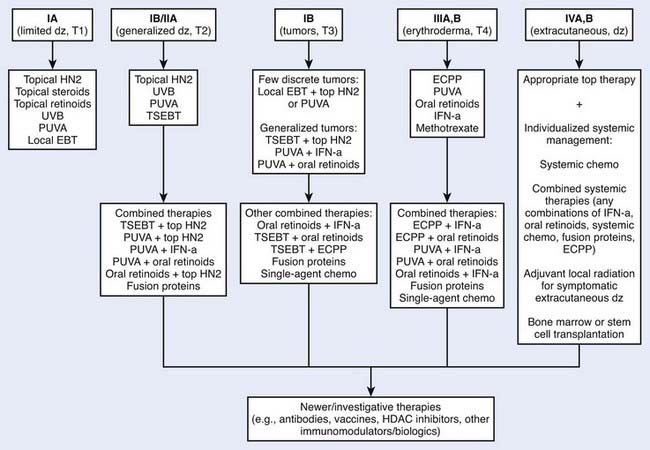
Figure 62-2 • An algorithm summarizing management approaches for patients with all stages of mycosis fungoides.
Topical Chemotherapy
Nearly all patients with mycosis fungoides require treatment directed at their skin. Common therapies include topical corticosteroids, psoralen plus ultraviolet (UV) A (PUVA), UVB, topical chemotherapy, topical retinoids, and irradiation. Topical nitrogen mustard (mechlorethamine, mustargen) is a very effective form of topical chemotherapy for patients with mycosis fungoides.30,35,36 The activity of intravenous nitrogen mustard as an alkylating agent for the management of systemic malignancy is well documented. The mechanism of action when nitrogen mustard is applied topically for the management of mycosis fungoides is less certain and may not be related simply to its alkylating agent properties. Its activity may be mediated by immune mechanisms or by interaction with the epidermal cell–Langerhans cell–T cell axis. It may be mixed in water or in an ointment base.
Because of its efficacy and ease of application, topical nitrogen mustard is employed widely as primary or secondary therapy in the management of patients with mycosis fungoides, especially those who have a limited or generalized patch or plaque phase of skin involvement (see Fig. 62-2). In patients with a discrete number of refractory lesions, treatment may be supplemented with local irradiation.37
Topical Retinoids
Bexarotene (Targretin) 1% gel, a retinoid X receptor–selective synthetic retinoid, may be an effective agent for the topical treatment of mycosis fungoides. Bexarotene gel is applied with a thin application to the patches or plaques as often as twice daily. Because of the irritant effect of the retinoids, it is not suitable for generalized application, but is used only when there are a discrete number of patches or plaques. A phase III trial of bexarotene included 50 patients with refractory or persistent early stage disease (stages IA to IIA).38 Responses were seen in 62% of patients with stage IA and 50% of patients with stage IB disease. Three patients with stage IIA or IIB disease did not respond. The most common toxicity of bexarotene gel is irritation at the sites of application, which occurs in the majority of patients. Because of the erythema from the irritant reaction, it may be necessary to withhold therapy for a few weeks to assess disease activity. Bexarotene gel is approved by the Food and Drug Administration (FDA) for patients with stages IA and IB disease who have refractory or persistent disease after other therapies or who have not tolerated other therapies.
Phototherapy
Phototherapy involves using UV radiation in the form of UVA or UVB wavelengths, which can be used alone, together, or with psoralen, a photosensitizing agent, as PUVA. The long-wave UVA has the advantage over UVB in its greater depth of penetration into the dermal infiltrates of mycosis fungoides. For early limited diseases, UVB alone39 or home UV phototherapy (UVA + UVB)40 has been shown to be effective.
PUVA is the most commonly used form of phototherapy for mycosis fungoides and Sézary syndrome patients. It was first used in the treatment of psoriasis but has proved effective for patients with mycosis fungoides as well.41–43 In the presence of UVA, the psoralen drug intercalates with deoxyribonucleic acid (DNA), forming both monofunctional and bifunctional adducts, which inhibit DNA synthesis.
Indications for PUVA treatment include its use as the primary therapy for patients with limited or generalized patch or plaque disease or as a secondary therapy following the failure of other topical modalities. PUVA may also be effective for patients with erythroderma, provided that very low daily exposures are used to avoid phototoxic reactions. For patients with Sézary syndrome, PUVA may be supplemented by systemic biologic therapies, such as IFN-alpha or systemic retinoids.44,45 The treatment of patients with more advanced cutaneous disease may be facilitated by the addition of localized irradiation to particular refractory plaques or tumors.
The primary acute complications of PUVA therapy include nausea and phototoxic reactions such as erythroderma and blistering, as well as skin dryness. Patients should shield their skin and eyes from the sun for at least 24 hours following psoralen ingestion. The potential long-term complications of PUVA therapy include cataract formation (requiring the use of UVA-opaque goggles during therapy) and secondary cutaneous malignancies. Among patients treated for mycosis fungoides, this risk is greatest for patients who have undergone long-term treatment with multiple topical therapies.46
PUVA may often be used in combination with other therapies, such as IFN or retinoids. For the combined regimen with PUVA, IFN-alpha and PUVA therapy are initiated concurrently, each usually given thrice weekly. When skin clearance is complete, the frequency of PUVA treatment is reduced. Complete and partial response (PR) rates of 79% to 80% and 14% to 20% in patients with generalized patch and plaque (stages IB and IIA) disease treated with the combination of IFN-alpha and PUVA have been reported.44 The clinical response and response duration results may be better with the combined regimen of PUVA and IFN-alpha compared with either treatment alone; however, prospective randomized clinical trials are needed to confirm this impression.47,48
When retinoids are used in combination with PUVA (RePUVA), the response rate is similar to that of PUVA alone; however, the responses may be achieved with fewer PUVA treatments and with a lower cumulative UVA dose.45 The duration of remission may be more prolonged if retinoids are given as maintenance therapy.49