Fig. 4.1
Strategies to identify mutant epitopes from transformed cells, CSF: cerebrospinal fluid
4.2 Antigen Processing and Presentation in Cancer
In order to gain an understanding of the dynamics driving the generation and ‘visibility’ of antigens to the immune system, it is advantageous to provide an overview about antigen processing and presentation to immune effector cells. Antigens can be generally viewed as being either intrinsic or extrinsic in nature; they are biochemically processed within cells and presented to various T-cell subsets, B cells as well as natural killer (NK) cells [8]. The essential molecule associated with presenting antigens to the immune system is termed as the major histocompatibility complex (MHC), or specifically in humans, the human leukocyte antigen (HLA) [8]. The function of MHC/HLA system was discovered and first described by Rolf Zinkernagel and Peter Doherty in the early 1970s, the seminal work for which they were awarded the Nobel prize in Medicine and Physiology in 1996 [9–12].
Antigens can derive from whole pathogens, i.e. bacteria, viruses and parasitic organisms, or by non-mutant, or mutant proteins associated by transformed cells. Intrinsic antigens, also called ‘endogenous’ antigens, are processed and presented to the immune system in the form of specific peptides called epitopes. This pathway is termed the MHC/HLA class I pathway (hereafter referred to as the ‘HLA class I pathway’), and plays a crucial role in eliciting immune responses to viruses (viral components synthesised within the host cell), intracellular bacteria as well as to cancer – associated antigens – which relies on the immune system’s capacity to recognized ‘self’ or ‘mutant self’ antigens [8]. All cells of the body (with the exception of erythrocytes) are capable of processing and presenting antigens via the HLA class I pathway. The processing of antigens in this pathway involves a crucial step, where the immunoproteasome (occurring in the cytosol) cuts up denatured (unfolded) protein structures into small peptide sequences between 8 and 10 amino acids long. The amino acid junctions at which the proteasome enzymatically cuts a protein decides on which peptide or epitopes are naturally presented to immune cells. Epitopes presented by HLA class I molecules are recognised by CD8+ T-cells, which can respond by i) proliferation, ii) cytokine production and / or iii) production of cytotoxic molecules, capable of killing transformed cells [8]. CD8+ T-cells may produce perforin, granzymes, and granulolysin (that can be easily measured using an CD107a induction assay), or – not mutually exclusive – IFN-gamma in response to transformed cells [13]. If (cancer) target epitopes are identified using the ‘reverse immunology strategy’, i.e. that epitopes are selected based on their predicted capacity to bind to MHC class I or class II molecules, it cannot be assumed with a very high degree of certainty that T-cells will also recognise the naturally processed and presented epitopes on tumour cells—a scenario which was described more than two decades ago [14]: T-cells that were shown to be peptide specific were not able to react against naturally processed and presented peptides on tumor cells. One of the reasons driving this phenomenon is that the specialised, or ‘skewed’ antigen processing and presentation machinery in transformed cells may be different compared to professional and non-professional antigen presenting cells [15] that are responsible for activating and expanding antigen-reactive T-cells. Alternatively, epitopes may have been created via post-translational modifications (such as phosphorylation) that could not be predicted from the primary structure of the wildtype and/or the mutant protein [16].
Antigens that are taken up from the external environment by professional antigen presenting cells or APCs (i.e. dendritic cells, macrophages), including B-cells, that have also professional APC functions, are usually processed and presented to the immune system via the HLA class II pathway. Whole pathogens, as well as proteins, e.g. generated via destruction of cancer cells by antibody-mediated mechanism, NK or CD8 T-cells, are actively taken up by APCs in endocytic vesicles called phagosomes, after which proteolytic enzymes contained within lysosomal compartments fuse with the phagosome to digest the antigen to yield smaller peptide sequences, usually 13–17 amino acids in length. These epitopes are then presented to CD4+ T-cells, which are also termed as helper T-cells (Th), and have an indispensable role in orchestrating immune responses mainly by producing effector cytokines, i.e. IFN-γ, tumor necrosis factor alpha (TNF-α), interleukin (IL)-2 (Th1 cells), IL-4, IL-10 (Th2 cells) and in some cases, IL-17 (Th17 cells). Cytotoxic activity is not exclusively attributed to CD8+ CTLs; cytotoxic CD4+ T-cells have also been reported to mediate biologically relevant immune responses in cancer as well as in viral infections [17–19].
The T-cell receptor (TCR) on the surface of T-cells binds to the HLA-epitope complex, along with co-receptors CD8 or CD4, to initiate an immune synapse. Interactions between T-cells and tumor cells are governed by HLA-restriction—the alleles encoding a person’s HLA repertoire and matching TCRs available in the tissue microenvironment and/or in blood, which dictates the nature and strength of the immune response. HLA allele-restriction of epitopes and immune cross-reactivity thereof plays an indispensable role in dictating the nature of immune responses. For example, HLA-DQ variants have been associated with increased susceptibility to certain infectious diseases; mutations in the β57 subunit of HLA-DQ may perpetrate progression to pulmonary disease [20]. Interestingly, mutations in HLA-DQ alleles have been attributed to susceptibility to contract type 1 diabetes mellitus (T1DM). While HLA-DQ is highly prevalent among Caucasians in the Americas as well as Europe, East Asians and Africans are much less likely to express these alleles [21]. Indeed, individual HLA alleles may also favour certain immune-recognition profiles, independent of the peptide repertoire displayed by the nominal restricting MHC element, i.e. HLA-DQ0602 favours IL-17 production independent of binding peptides, as shown in the transgenic murine model of multiple sclerosis [22]. This IL-17-centric reactivity represents a double-edged sword; it may more effectively contain certain bacterial infections [23, 24] and IL-17 may be beneficial to attract immune cells to the tumor site [25] while the chronic exposure to IL-17 may rather promote malignant transformation [26–28]. Therefore, the nature, quality and quantity of immune responses following vaccination appear to greatly depend on an individual’s HLA profile, which shapes the quality and quantity of ensuing cellular immune responses, including increased or decreased risk for infections, autoimmune responses or the ability to present (neo) epitopes to T-cells dependent on the restrictions imposed by the MHC–peptide complex and the responding TCR repertoire. For instance, even if neoepitopes are generated during malignant transformation, they may not be visible to the cellular immune system, if they are not processed and ultimately complexed to the respective HLA molecule and presented to responding T-cells.
Th1–Th2 Responses and MHC Restriction
Most studies use IFN-gamma as the readout of T-cells responding to wildtype and mutant epitopes provided from cancer cells, yet Th2 responses, with the signature cytokines IL-4, IL-5 and IL-13 may also be present, either as an ‘original’ Th2 response or as a result of partial agonist peptides, imposed by the mutational event (see below) that may turn Th1 T-cells into Th2 cytokine-producing T-cells [29]. Th2-type T-cell responses may not per se signify an unproductive and potentially ‘tolerizing’ immune response; more recent reports indicate that Th2-type immune responses may also be able to mediate clinically relevant anti-cancer immune reactivities [30]. In a preclinical model, antigen-specific Th2 cells eradicated myelomas without the help of CD8 T-cells, leading to massive inflammation at the tumor site [30]. Th2-mediated tumour destruction has been shown to be associated with IL-1, TNF-alpha (Th1) and Th2 cytokine (IL-4, IL-5, IL-13) production in situ, while passively transferred Th2 cells were able to confer long-lasting cellular anti-cancer directed immune responses. CD8-independent and antigen-specific T-cells in Th2-mediated immune responses were shown to be eotaxin- and STAT6-dependent [31–36]. In general, Th2 infiltrates in human cancers have not been studied extensively and some studies even suggested a better outcome with Th2-type cytokines [36]. The nature of Th2 responses in recognising mutant epitopes is not well explored at this time. The more detailed association of CD4 Th2 responses may also benefit from closer association of T-cells with the restricting MHC class II elements. For instance, previous studies reported Th1/Th2 CD4+ T-cell responses against NY-ESO-1 in DPB1*0401/0402-positive patients with ovarian cancer [37]. Much more information is available concerning the nature of the cellular immune response directed against peptides presented by the rather less variant (as compared to HLA-DR) HLA-DP molecules from infectious pathogens, e.g. Hepatitis B or MHC class II molecules that pre-dispose humans to certain autoimmune diseases (e.g. gluten-associated colitis) [37–45]. The impact of variant epitopes in association with certain MHC alleles that are associated with certain cytokine production patterns (IL-17, Th1, Th2) is unexplored up to now. Table 4.1 provides an overview of wildtype and mutant target epitopes recognised in TIL from patients with glioma, demonstrating that Th2 responses exist in the TCR repertoire from individual patients directed against mutant epitopes.
Table 4.1
TIL reacting against wildtype and mutant epitopes—Th1 and Th2 patterns
ID | Patient code | Wildtype sequence | IFN-γ wildtype | IFN-γ mutant | TNF-α wildtype | TNF-α mutant | IL-17A wildtype | IL-17A mutant | IL-4 wildtype | IL-4 mutant | IL-5 wildtype | IL-5 mutant | IL-13 wildtype | IL-13 mutant | Mutated sequence | Gene ID |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
1 | GBM-1 | ALYDICSKTLKLPTP | 193.09 | 570.95 | 64.68 | 22.69 | 11.75 | 36.71 | 45.35 | 33.71 | ALYDICSRTLKLPTP | TUBB8 | ||||
2 | GBM-1 | SSGGCCGSSSGGCCS | 63.38 | 4.92 | 9.07 | 3.96 | SSGGCCGSSSGGCCS | LCE1F | ||||||||
3 | GBM-1 | AKQTSNCVLEICAEQ | 17.37 | 4.74 | 2.31 | AKQTSNCILEICAEQ | ESPNP | |||||||||
4 | GBM-1 | REQEEKMWRQEEKIR | 126.84 | 85.39 | 3.94 | 30.39 | 4.95 | 11.85 | REQEEKMRRQEEKIR | NCKAP1L | ||||||
5 | GBM-1 | REDAGAGGEDVGAGG | 88.74 | 37.28 | 1.98 | REDAGAGEEDVGAGG | GOLGA6L1 | |||||||||
6 | GBM-1 | IREQEEMLREQEAQR | 105.48 | 754.23 | 35.73 | 18.34 | 53.61 | IREQEEMiREQEAQR | GOLGA6L2 | |||||||
7 | GBM-1 | PPTWSGRRAPGDRDN | 270.84 | 69.22 | 19.06 | PPTWSGRHAPGDRDN | LOC645752 | |||||||||
8 | GBM-1 | QFLIPTSLSVSSNSV | 30.48 | QFLIPTSFSVSSNSV | DSPP | |||||||||||
9 | GBM-2 | WPSFSEAHGTSGSDE | 428.21 | 747.66 | 76.27 | 86.03 | 28.36 | WPSFSEARGTSGSDE | MSRB2 | |||||||
10 | GBM-2 | TATASSTQATAGTPH | 196.06 | TATASSTRATAGTPH | MUC5B | |||||||||||
11 | GBM-2 | TATATTTGATGSVAT | 31.21 | TATATTTRATGSVAT | MUC5B | |||||||||||
12 | GBM-2 | NLKEKCFLTQLAGFL | 9.24 | NLKEKCFVTQLAGFL | NBPF8 | |||||||||||
13 | GBM-2 | LLTPDEPDKSQGQDL | 18.28 | 3.1 | 116.92 | LLTPDEPEKSQGQDL | NBPF12 | |||||||||
14 | GBM-2 | PDAVGKCRSAGIKVI | 353.94 | 865.82 | 15.74 | 60.5 | 36.45 | PDAVGKCGSAGIKVI | ATP1A2 | |||||||
15 | GBM-2 | ARCSSEDDDSKESTC | 412.16 | 47.28 | 7.53 | 147.1 | 26.11 | ARCSSEDSKESTCSP | PHOX2A | |||||||
16 | GBM-2 | RWEEWNKRLEEVKRE | 30.69 | RWEEWNK | AK7 | |||||||||||
17 | GBM-2 | PGEGHGGEHLDSEGE | 7.41 | PGEGHGGDHLDSEGE | GOLGA8DP | |||||||||||
18 | GBM-2 | PSDLRRHVRTHTGEK | 81.47 | 24.61 | 42.07 | 30.96 | PSDLRRHMRTHTGEK | ZNF764 | ||||||||
19 | GBM-2 | EGGPAAPRLGSRTAP | 288.05 | EGGPAAPHLGSRTAP | LINC00273 | |||||||||||
20 | GBM-2 | NRPTSGPWQRHTRRS | 495.14 | 19.38 | NRPTSGPRQRHTRRS | LINC00273 | ||||||||||
21 | GBM-2 | ADPIPSGLSPGPCGA | 31.21 | ADPIPSGRSPGPCGA | LINC00273 | |||||||||||
22 | GBM-2 | MKDCQLRKQQNENVS | 41.12 | MKDCQLRRQQNENVS | SLFN12L | |||||||||||
23 | GBM-2 | VKRNPPKTAKVSEPG | 246.07 | 1275.82 | 9.24 | 60.58 | 84.52 | 169.86 | 49.05 | VKRNPPKKAKVSEPG | HOXB1 | |||||
24 | GBM-2 | SAFEPEGVLANVLGL | 65.93 | 25.85 | SAFEPEGGLANVLGL | CYB561 | ||||||||||
25 | GBM-2 | GSGPSCRRWEKKLAS | 376.6 | 25.49 | GSGPSCRQWEKKLAS | PIK3R5 | ||||||||||
26 | GBM-2 | DMYGTGQESLYS | 472.45 | 69.15 | 31.08 | DMYGTGQQSLYS | CDH7 | |||||||||
27 | GBM-2 | QSYKNDFNAEYSEYR | 385.13 | 37.04 | QSYKNDFSAEYSEYR | ELL | ||||||||||
28 | GBM-2 | ARKAKYNVHATVRYQ | 376.14 | 3.01 | ARKAKYNLHATVRYQ | NCAN | ||||||||||
29 | GBM-2 | MRVMKFSVSPVVRVA | 258.99 | MRVMKFSISPVVRVA | EEF2 | |||||||||||
30 | GBM-2 | YAPCGDLSGMLQERG | 23.08 | YAPCGDLNGMLQERG | SBK3 | |||||||||||
31 | GBM-2 | GQLAVSKRLALEVTV | 304.12 | 39.39 | GQLAVSKCLALEVTV | SIRPG | ||||||||||
32 | GBM-2 | QRAAAIARQKAEIAA | 889.28 | 112.28 | QRAAAIACQKAEIAA | JPH2 | ||||||||||
33 | GBM-2 | ISPSRAARQLMERTQ | 606.87 | 120.34 | ISPSRAACQLMERTQ | ELMO2 | ||||||||||
34 | GBM-2 | FARKLKDVHETLGFP | 410.66 | FARKLKDIHETLGFP | TTN | |||||||||||
35 | GBM-2 | EPDNIKYVISEEKGS | 361.16 | EPDNIKYMISEEKGS | TTN | |||||||||||
36 | GBM-2 | DNHCEQLRVKIRKLK | >1864.49 | >1864.49 | 184.5 | DNHCEQLGVKIRKLK | ANKRD36C | |||||||||
37 | GBM-2 | LTELKDNHCEQLRVK | 495.5 | 561.95 | 1759.72 | 80.89 | 34.27 | LTELKDNLCEQLRVK | ANKRD36C | |||||||
38 | GBM-2 | DFSVIIMAYVSENIK | 669.42 | DFSVIIMVYVSENIK | SCN5A | |||||||||||
39 | GBM-2 | GKGVMLAVSQGRVQT | 328.34 | 676.71 | 702.12 | 69.23 | 53.48 | GKGVMLAISQGRVQT | TENM3 | |||||||
40 | GBM-2 | ESRGLLQRRAQAAQE | 214.08 | ESRGLLQHRAQAAQE | CFAP99 | |||||||||||
41 | GBM-2 | SRELCPGRWRAGPWS | 467.9 | 1023.03 | 52.66 | SRELCPGHWRAGPWS | ADAMTS2 | |||||||||
42 | GBM-2 | WDLTDALRLAALSIE | 423.55 | WDLTDALWLAALSIE | LAMA4 | |||||||||||
43 | GBM-2 | SHLIAASNCHSLELQ | 993.08 | 146.98 | 33.34 | SHLIAASSCHSLELQ | CAPN11 | |||||||||
44 | GBM-2 | EDVKWPPTLQPPTLR | 682.55 | EDVKWPPPTLQPPTL | IRF5 | |||||||||||
45 | GBM-2 | NGMEWNGMEWNRIES | 343.39 | NGMEWNGIEWNRIES | PCSK5 | |||||||||||
46 | GBM-2 | AATSHPKPTTGHIKP | 307.31 | 1000.95 | 14.03 | 134.72 | 109.23 | 33.29 | AATSHPKHIKPATSH | GPR50 |
Processing and presentation of neoantigens may yield mutant epitopes (neoepitopes) that are shared as well as patient-specific (‘private’). This of course depends on the location of the mutation, i.e. point mutation which might disrupt the naturally occurring cleavage site and the nature of the mutation itself i.e. point mutation vs. chromosomal deletion vs. premature stop codons. A comprehensive analysis of somatic mutations in the HLA class I pathway, using DNA isolated from tumour and non-tumour tissue from patients representing 20 different cancer types, revealed a high likelihood for loss-of-function mutations occurring in the N-terminus of the HLA class I molecule, which abrogates transport of the peptide-HLA complex to the cell surface [46]. Furthermore, in all cancers tested, the most frequent mutations were found to occur in the α3 region of the HLA class I molecule, which is required for binding of the CD8 co-receptor on T-cells during an immune synapse for subsequent activation of the CD8- TCR complex [8].
4.3 Cancer Antigens and Epitopes: From Discovery to Therapeutic Application
Preclinical studies in the mouse model of human cancer, in particular melanoma, provided the first insights into cancer antigen discovery and functional characterisation, in the context of tumour rejection. Thierry Boon and colleagues had shown in the late 1980s that the tumor– antigen P19A, heterologously expressed in mouse P815 tumour cells (isolated from DBA/2 mice bearing methylcholanthrene-induced sarcoma), contains an HLA class I epitope (within a 13-mer sequence harbouring a point mutation) capable of eliciting potent CTL responses and lysis of target cells [47].
Epitope mining in the human cancer setting was first performed using tumour tissue derived from human melanoma lesions, spearheaded by groups in Europe and the United States. Thierry Boon, Pierre Coulie and colleagues at the Ludwig Institute in Brussels, Belgium discovered the first tumour-associated antigen (TAA) in 1991, after in vitro characterisation of CTL responses using melanoma cell lines derived from an anonymous patient MZ2 who had metastatic disease [48]. This TAA, first annotated as MZ2-E and later renamed as melanoma-associated antigen 1 (MAGE-1, cancer testis antigen), was recognised by an autologous CTL line and induced lysis of the tumour cell line expressing the MAGE-1 DNA and restricted by HLA-A1 [48]. Further work with a cell line from the same patient led to the discovery of MZ2-F, or as it is known today, G antigen 1 (GAGE-1) [49]. Much of the ongoing work at the time focussed on discovering novel immunogenic HLA class I-restricted antigens that mediated CTL reactivity and lysis of melanoma cells from patients, with a strong interest to first understand and then to develop immune-based interventions; Melan-A (HLA-A2+ epitope) [50]; MAGE-3 (HLA-A1+ epitope)-specific CTL response in a patient vaccinated with MAGE-3.A1 peptide [51].
Simultaneous efforts by researchers in Europe and the United States revealed another important cancer antigen, the cancer testis antigen NY-ESO-1, which was discovered by serological analysis of expression cDNA libraries (SEREX) (indicating the presence of antibody responses), using cDNA prepared from human oesophageal squamous carcinoma cells [52]. NY-ESO-1 was later shown by Elke Jäger and co-workers (Frankfurt) to contain biologically functional CD8+ (HLA-A2/B51) and CD4+ (HLA-DRB*1) T-cell epitopes, based on seminal studies performed on human melanoma cells as well as transfected T2 cells as a model [53–56]. The afore-mentioned T2 cells harbour a defect in the transporter associated with antigen processing (TAP), which in turn inhibits them to present endogenous cytosolic cytosolic peptides (except for some leader peptide sequences loaded onto HLA-A2 molecules), but accommodates the introduction of exogenously added HLA class I epitopes for CTL recognition assays [57].
Steven Rosenberg and colleagues at the Surgery Branch, National Cancer Institute (NCI, National Institutes of Health (NIH), Bethesda, MD) made pivotal contributions to antigen discovery in human melanoma, in particular those that induce reactivity among TILs: the tyrosine related protein 1 (TRP-1) or gp75 restricted by the HLA-A31 molecule in 1995 [58]; HLA-A31-restricted TRP-2 peptide LLPGGRPYR, which was a major target of TILs infused into a patient with metastatic melanoma who thereafter showed disease regression [58]; epitopes from TRP-1 and TRP-2 (TRP197–205) restricted by HLA-A31 as well as HLA-A33 [59]; a mutated epitope derived from triosephosphate isomerase restricted by HLA-DR1 and recognised by CD4+ TIL and cell division cycle protein 27 homolog (CDC27) epitope restricted by HLA-DR4 [60, 61]. Collectively, these early efforts (over a span of 15 years, from the late 1980s to early 2000s) provided an excellent foundation which lead to the expansion of the field of targeted cancer immunotherapy.
A whole series of other molecules were identified to be associated with transformed cells. For instance, mesothelin was discovered as a marker of several important solid cancers, i.e. mesothelioma, ovarian cancer, pancreatic ductal adenocarcinoma based on serological (a murine ‘Ki antibody’ recognising human mesothelin) and genetic analyses [62–64]. Further exploration of the clinical significance of this molecule in ovarian cancer, mesothelioma and squamous cell carcinomas, and in conjunction with measurable mesothelin as well as antibody responses in sera of patients, indicated the immunogenic potential of mesothelin and its designation as a legitimate cancer antigen [65, 66]. An experimental immunotoxin developed based on the mesothelin-binding region of the K1 antibody was among the earliest attempted targeted immune-based interventions, with preclinical studies performed in a murine model of human carcinoma xenografts [67].
Work implemented in the later part of the 1990s placed a greater focus on studying mutated proteins in human cancer cells, and the possibility of discovering mutated antigenic determinants (neoepitopes) presented by HLA restricting elements, with biological and clinical relevance in therapy. An early example is a neoepitope derived from melanoma ubiquitous mutated 1 protein (MUM-1, initially named LB33-B, after the patient from whom the melanoma tumour was obtained, LB33 [68]), which is restricted by the HLA-B*44*02 allele. This 9-mer neoepitope was identified following in vitro cytotoxicity studies directed against the autologous melanoma cell line LB33-MEL.A-1; the same cytolytic activity was not seen with the wildtype peptide sequence [69]. A 10-mer neoepitope (amino acids 23–32) from mutated cyclin-dependent kinase 4 (CDK4R24C) protein, restricted by HLA-A*0201, was also shown to mediate cytolytic activity by autologous CTLs in a dose-dependent manner, when exposed to T2 cells transfected with the CDK4R24C cDNA [70]. A caspase 8-derived mutated peptide restricted by HLA-B*3503, which showed potent cytolytic activity against the autologous head and neck cancer cells as well as tumour cDNA-transfected B-cell lines [71] further strengthened the field of neoepitope mining from human cancer cells.
A high-throughput analysis of whole genomic as well as exomic DNA from clinical tumor samples representing thirty different human cancers revealed the unique mutational burden in each cancer type, in addition to specific mutational signatures characterising these cancers [72]. Although this provides an elegant view of the general landscape of mutational burden in human cancers, the mutational signature in each patient varies—thus giving rise to a ‘compendium’ of private mutational signatures involved not only in driving and maintaining malignant transformation, but also in the activation and expansion of immune effector cells.
The mutated form of the V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog, or known as KRAS in short, is a well-established neoantigen implicated in the pathogenesis of pancreatic, colorectal and lung cancers [73–76]. Native KRAS was discovered in 1982 following gene sequencing of human lung adenocarcinomas, and is a guanine triphosphatase involved in cellular signal transduction [77]; however, mutations at positions 12, 13 and 16 are associated with oncogenesis, thus making it a proto-oncogene in humans.
Steven Rosenberg and colleagues at the Surgery Branch, National Institutes of Health (Bethesda, MD) recently developed a cutting-edge approach to screen for neoepitope-specific T-cell responses for individual patients. This method has been termed the ‘tandem minigene (TMG)’ approach, which first requires whole-exome sequencing data of genomic DNA isolated from patients’ tumor tissue samples. The sequencing data then yield all non-somatic mutations contained within gene-coding DNA of the patient. This allows for constructing a personalised library of the patient’s ‘private’ mutations that potentially code for neoepitopes. These short gene sequences are then put together, an artificial construct, and inserted into an expression plasmid, which is subsequently transfected into a lentiviral vector for infection of APCs, i.e. dendritic cells (DCs) from a patient. Autologous TILs (from the same patient) are then co-cultured with the TMG-bearing DCs to allow induction of immune-reactivity. A positive response, represented by IFN-γ production by the TILs, would signal that the co-cultured DCs harbour a TMG that includes a neoepitope-encoding sequence(s) that is/are naturally processed and presented to the immune system [78].
Mutations may lead to different, not mutually exclusive effects on the responding T-cell population based on the prerequisite that a mutant epitope is indeed processed and presented to T-cells: (1) a T-cell may be newly recruited that would exclusively recognize the mutant sequence, (2) potential T-cell receptor (TCR) cross-reactivity between wildtype and mutant epitope sequences (if both, the wildtype and the mutant epitopes are being processed and presented to T-cells). It is biologically relevant whether there is already a T-cell population expanded that recognizes the wildtype epitope and then, following malignant transformation, recognizes the mtuant target, since this situation may lead to differential TCR triggering and subsequently to differential T-cell effector functions.
Mutant Epitopes as the ‘Biological Scalpel’ Against Cancer Cells
Increasing immune effector functions by recruiting T-cells that recognise mutant epitope sequences is a clinically attractive attempt to improve and broaden the TCR repertoire directed against mutations that exclusively exist in malignancies and would therefore represent the ‘ideal’ cancer-associated antigen – a ‘biological scalpel’ that would only target cancer cells and not harm non-transformed cells. One method is to modify peptides at residues that do not interact with the nominal MHC restricting molecule, yet with the TCR contact residues: these variants are called ‘heteroclitic’ analogues and are able to trigger the nominal TCR with differential T-cell effector functions, e.g. cytotoxicity, quality and quantity of cytokine production, as well as proliferation [79]. This approach has been used to induce T-cells that react to wildtype peptides, e.g. to p53, yet are elicited with a variant peptide. If single mutations occur in epitopes, it could yield peptides that are naturally processed and presented by tumour cells to TCRs. What could potentially happen if a T-cell response, directed against a wildtype target, will also be able to able to react to the corresponding mutant target epitope, presented by the identical, nominal HLA-restricting element?. The following scenarios may occur, which have been described already in the early 1990s from several groups: In general, a single ligand specificity for each individual TCR appears to be rare. In contrast, the TCR recognition has been shown to be flexible, induced by altered peptide ligands, grouped into antagonists, partial agonists and superagonists. Mutations within peptide targets can induce differential phosphorylation of the TCR/CD3 complex with a differential downstream signalling pathway configuration [80]. Mutations may therefore—in case the wildtype peptide is also recognized—lead to abrogation of T-cell recognition simply because the ligand is not processed and presented. Alternatively, the T-cell ligand may well be processed and presented, but the T-cell signal may be abrogated potentially due to cellular anergy [29]. Partial agonists, i.e. by inducing a single amino acid residue, will still be able to stimulate the T-cells directed against the wildtype peptide, even across a similar dose range of the nominal epitopes: as the T-cells with the wildtype TCR ligand react with proliferation, cytokine production and cytotoxicity, peptide variants may either induce cytotoxicity and/or cytokine production, in the absence of T-cell proliferation [81, 82]. Of note, a similar observation may be true for TCRs directed against the mutant epitope that would react with a qualitatively and quantitatively differential T-cell reactivity pattern. Single amino acid exchanges may also turn T-cell clones from a Th1 into a Th2 cytokine production pattern, or lead to T-cell clones with abrogated cytokine production, yet strong cytotoxic T-cell responses as shown for viral pathogens [81–83]. Mutations in nominal targets, associated with differential signalling events, may also be crucial for the differentiation status of T-cells reacting to wildtype as well as to mutant targets, first described in preclinical models of thymocyte differentiation and maturation [84, 85]. Differential triggering of the nominal TCR is associated with T-cell maturation and differentiation—a quality that is important for long-term immune memory, access to (tumor) tissue as well as for clinical efficacy of T-cell therapy. The passive transfer of immune cells directed to TAAs has been shown to be clinically relevant as the transfer of the T-cell product leads to the generation of central memory T-cells [86]. The biochemical signals that govern T-cell memory rely not only on the cytokine environment, yet also on the signal strength delivered by the TCRζ chain complex; the quality and quantity of T-cell responses including T-cell memory is strikingly shaped by the strength of the MHC/peptide–TCR interaction [87], which may in part be relevant for CD8+ T-cells. A decreasing potential model has been proposed, gauging the signal strength delivered by the target epitope to the corresponding TCR that is dictating whether the T-cell most likely enters the T-cell memory pool [88, 89]. This ‘signal strength’ model will need to take into account the locally produced cytokines and pro-inflammatory signals associated with moving T-cells into the diversity of the memory T-cell pool. In general, weaker TCR signals are sufficient in order to move T-cells into a memory T-cell program [90, 91]. Not mutually exclusive, the length of the TCR signalling (i.e. shortening the TCR stimulation) will also decide whether T-cells enter the memory T-cell pool [92, 93]. The observation that point mutations within peptides affect the contact with the nominal TCR also impairs CD8+ T-cell memory development, mediated in part by TCR-dependent NFκB signalling [94]. This may partly explain why T-cell clones targeting the identical (mutant) tumor epitope exist in heterogeneous differentiation states (see Fig. 4.2).
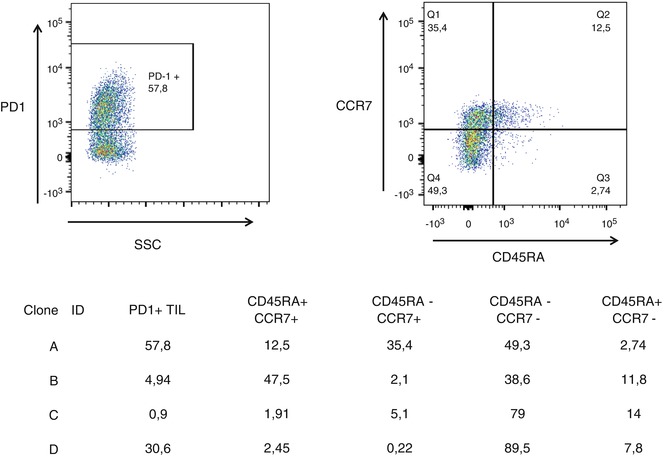
Fig. 4.2
Different T-cell clones (A: VB5.1, B-D: VB9) recognize a naturally processed and presented (mutant) target on autologous pancreatic cancer cells. Note that the cancer – directed T-cell clones expresses different T-cell homing and differentiation markers, defined by CD45RA and CCR7 expression, i.e. CD45RA+CCR7+ T-cells are precursor T-cells, CD45RA-CCR7+ central memory T-cells, CD45RA-CCR7- T-cells memory effector T-cells and CD45RA+CCR7- T-cells represent terminally differentiated effector T-cells. Note that the majority of cells derived from clone B reside in the central memory T-cell subset that has been shown to be associated with increased responsiveness in the cellular therapy of cancer
The role of gamma-delta (γδ) T-cells in cancer is now also being revisited, due to their non-classical recognition of antigens. γδ T-cells recognise non-peptide structures, i.e. phosphoantigens such as derivatives of the eukaryotic isoprenoid (mevalonate) pathway presented by the CD1d molecule [95]. The Vγ9Vδ2 (Vδ2+) subset of γδ T-cells, which are found in peripheral blood. They have also been described to express the CXCR3 surface marker which is crucial for tissue penetration, an important feature in accessing transformed cells or tissue-residing pathogens. An interesting feature of Vδ2+ γδ T-cells is that they express the CD16 co-receptor, which can bind to FcγRIII present on tumour cells in addition to the killer receptor NKG2A [96]. Thus, like NK cells, γδ T-cells can also orchestrate antibody-dependent cellular cytotoxicity (ADCC), which is implicated in the therapeutic activity of several monoclonal antibody-based cancer drugs, i.e. rituximab, trastuzumab, ofatumumab and alemtuzumab [97–99]. An intermediate of the isoprenoid pathway, isopentenyl pyrophosphate (IPP), is strongly recognised by Vγ9Vδ2 T-cells, as shown in the context of zoledronic acid-treated human cancer cells [100]. Zoledronic acid induces accumulation of IPP in cancer cells, thus stimulating the activity of Vγ9Vδ2 T-cells, subsequently promoting the production of IFN-γ as well as cytotoxic molecules. This effect can be further enhanced in the presence of IL-2 and/or IL-15 conditioning. Although altered/mutated forms of IPP are yet to be reported, the significance of γδ T-cells in targeted cellular therapy should be explored further. There have also been reports of the recognition and killing of overexpressed human heat shock protein 60/70 on cancer cells by γδ T-cells, indicating that the overall T-cell repertoire in human which recognizes tumor antigens is rather generous [101, 102].
4.4 Clinical Significance of Neoepitope-specific Immune Responses
The clinical value of neoantigen-specific responses is most evident in immune checkpoint blockade therapy. Case reports of patients with melanoma or non-small cell lung cancer treated with anti-PD-1 and anti-CTLA-4 monoclonal antibodies showed that the repertoire of neoantigen-directed CD8+ T-cell responses (based on the diversity of TCRs recognizing mutated peptides) is is associated with clinical responses [103–108]. The most relevant examples are the T-cell responses from patients with metastatic melanoma or non-small cell lung cancer NSCLC, whereby the number of PD-1+ circulating T-cells directed against neoepitopes (visualised by flow cytometry) associates with clinical outcome in patients [105, 109, 110]. Furthermore, more recent clinical observations indicate that neoepitope-specific immune responses in peripheral blood can be used as a prognostic marker for several solid cancers [107, 110, 111].
Tissue scarring, arising from inflammatory processes processes, associated with infection(s), may lead to to genetic aberrations, which in time may perpetrate oncogenesis. Observations in patients with lung adenocarcinomas who had previously contracted M. tuberculosis infection in the lung showed that immune responses to mycobacterial antigens (‘old’ tuberculosis (TB) lesions) caused mutational changes to the gene encoding epidermal growth factor receptor (EGFR), in association with cancer development [112]. More strikingly, these patients had a worse 1-year survival prognosis compared to those who did not have ‘old’ TB lesions in the lung at cancer diagnosis. Patients presenting with ‘old TB lesions’ and adenocarcinomas in the same lung did not harbour the EGFRL858R mutation (occurring in exon 21 of the EGFR gene, which encodes the intracellular tyrosine kinase domain of the receptor), which is implicated in positive clinical outcomes in patients with lung cancer who are treated with the tyrosine kinase inhibitor gefitinib [113]. The EGFRL858R mutation has been shown to give rise to neoepitopes that induce antibody responses in patients with NSCLC who received gefitinib therapy [114]. Another EGFR-associated mutation, EGFRT790M, which is found in approximately 60% of patients with NSCLC, yields HLA-A2-restricted neoepitopes that are linked to favourable anti-tumor immune responsesthat could be implemented for designing better immunotherapies [115, 116].
The agonistic activity of peptides, namely their ability to stimulate T-cell activation can in fact shape the cellular immune response milieu due to mutational changes in their molecular structure. Paul Allen and co-workers had elegantly shown in the mid-1990s that peptide analogues of staphylococcal enterotoxin A, derived from haemoglobin, can abrogate the effector functions while inhibiting the proliferation of T helper cells (CD4+ T-cells with a Th1 or Th2 phenotype) [117]. While some mutations in the haemoglobin peptides inhibited T-cell proliferation, other mutations did not have a deleterious effect on the T-cell. Further research showed that partial phosphorylation of the immunoreceptor tyrosine-based activation motif (ITAM), which forms an indispensable component of the intracellular TCR zeta (ζ) chain, can either totally abrogate or even lead to T-cell death during an immune synapse [118]. Importantly, this phenomenon can be due to the binding of TCR with HLA molecules presenting mutated peptides, and more importantly, the nature of the mutation itself and the very position of the mutation within the epitope sequence. It is undeniable that the local inflammatory milieu in cancer lesions (such as those described in chronic infections [119]) may also contribute to chromosomal aberrations resulting in strong downregulation or loss of the TCRζ chain. These seminal findings were first reported in a preclinical murine model of colon carcinoma and later in TILs from patients with renal cell carcinoma and peripheral blood T-cells from patients with non-Hodgkin’s lymphoma [120–122].
Preclinical studies of infectious disease models may provide an insight into TCR repertoire shaping in relation to neoepitope-specific immune responses. Analyses of splenic and bronchoalveolar lavage fluid-derived T-cells from mice primed with a wildtype strain of influenza A virus (HK/PR8) by intraperitoneal infection showed that animals’ CD8 TCRVβ repertoire was shaped by primary viral challenge to efficiently recognize and respond to a secondary challenge with another wildtype strain but not a mutated version of either virus (HK/PR8-NPN3A) [123]. Also, while challenge with a wildtype virus strain provided a broader TCRVβ repertoire, the mutant strain of the virus induces a more focussed and narrow antigen-specific T-cell compartment, with subtle TCR re-arrangement patterns. Furthermore, an immunogenic epitope from the wildtype virus (NP366, ASNENMETM) induced a measurable CD8+ T-cell response among mice primed and re-challenged with a mutated viral strain. Conversely, the mutated version of the NP366 epitope, harbouring only a single amino acid change (NPN3A366, ASAENMETM), did not promote strong binding between MHC and TCR among T-cells from mice challenged with a wildtype virus, exhibiting a high ‘off-rate’ (large percentage of mutated epitope-bearing tetramers dissociating from the TCR within minimal time), requiring greater dependence on the CD8 co-receptor binding to MHC to elicit an immune response. This is of relevance to immune responses in cancer; T-cell reactivity to neoepitopes may be subdued owing to poor binding kinetics between the HLA-restricting element and the TCR. However, vaccination with a broader array of personalised neoepitopes may help prime the immune system to either re-awaken the smaller populations of central memory T-cell that are tumor-reactive, or not mutually exclusive, generate a fresh pool of (as yet not activated) antigen-specific T-cells. [124].
Related posts:
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 Current Status of Immuno-Oncology in Hematologic Cancers
Current Status of Immuno-Oncology in Hematologic Cancers
 Synergy Between Radiotherapy and Immunotherapy
Synergy Between Radiotherapy and Immunotherapy
 Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
Toward Engineered Cells as Transformational and Broadly Available Medicines for the Treatment of Cancer
 Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
Plasmacytoid DC/Regulatory T Cell Interactions at the Center of an Immunosuppressive Network in Breast and Ovarian Tumors
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


