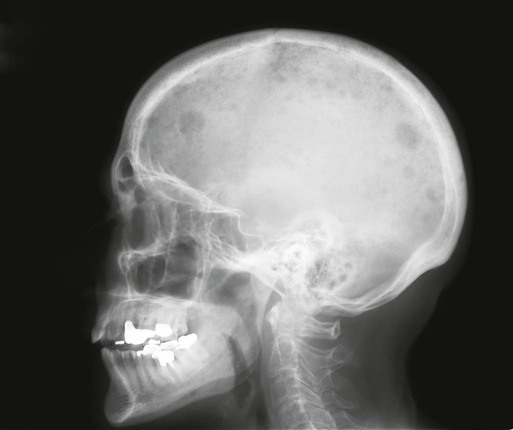
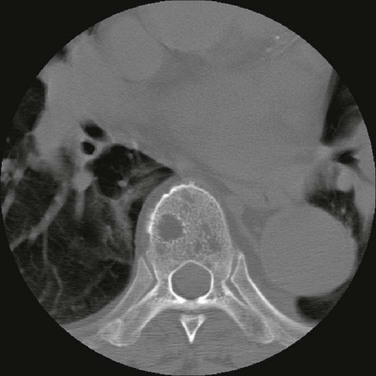
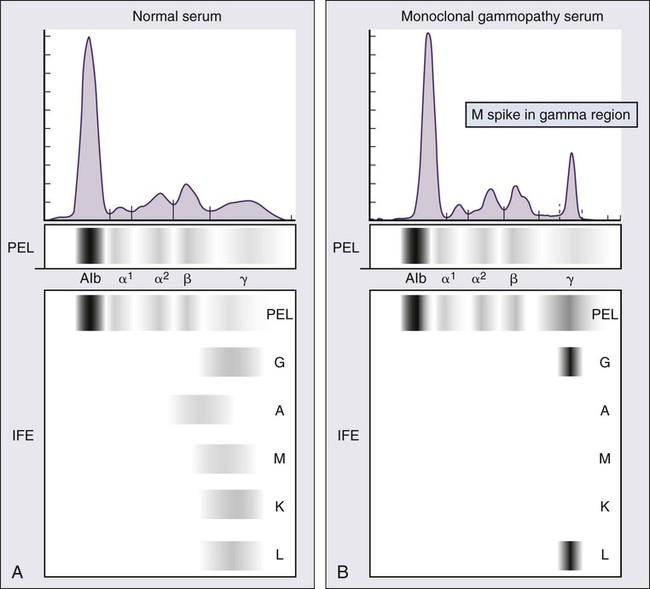
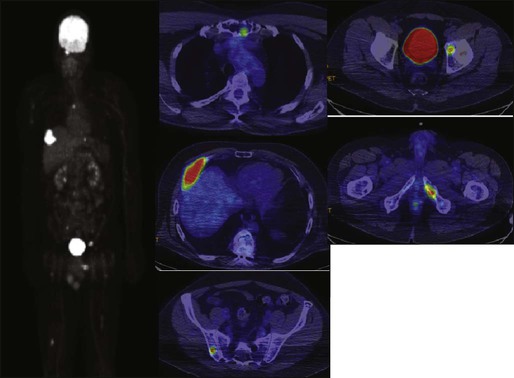
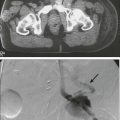
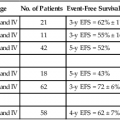
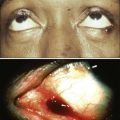
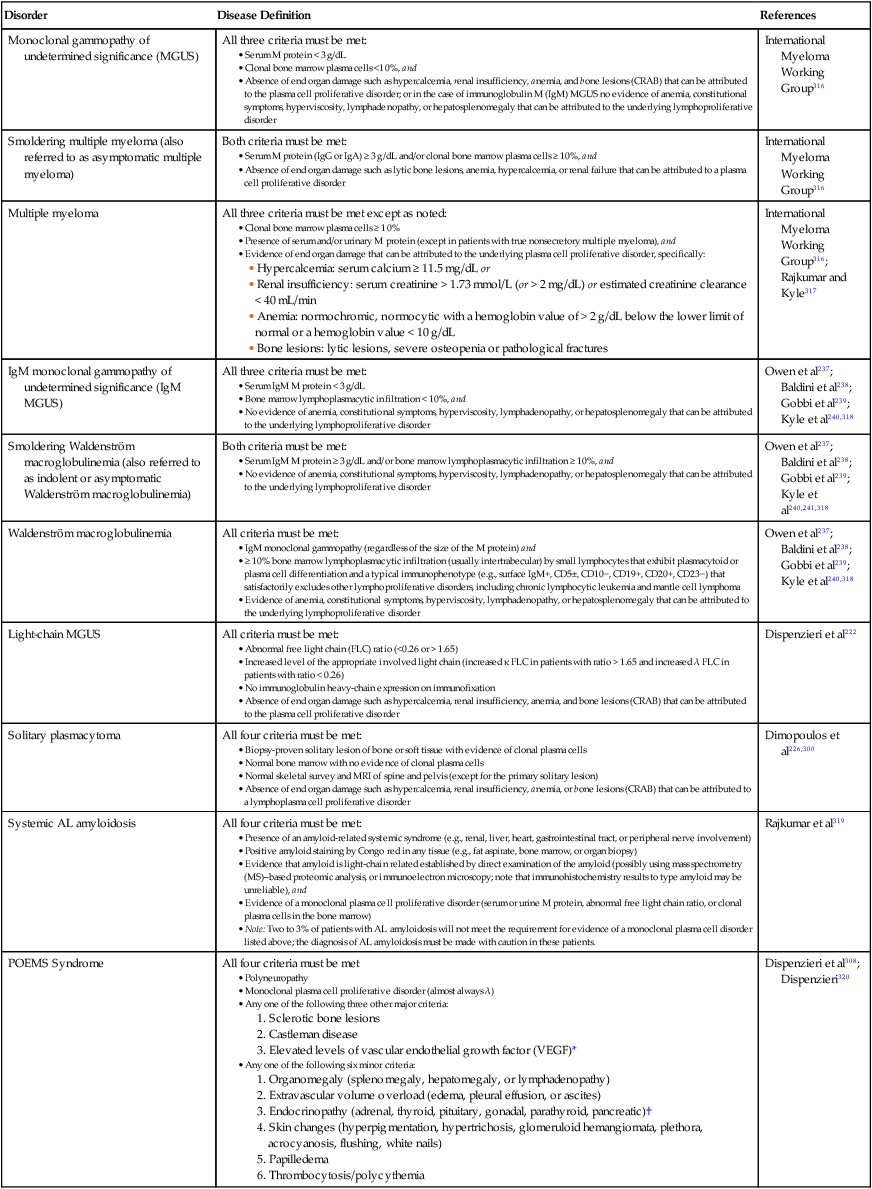
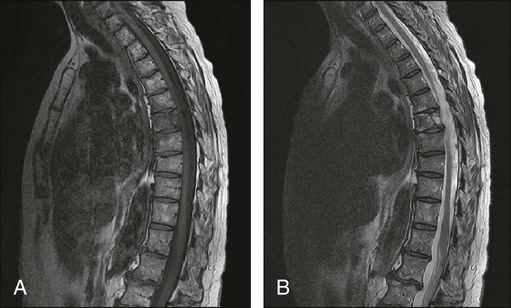
104 S. Vincent Rajkumar and Angela Dispenzieri • Multiple myeloma accounts for approximately 10% of hematologic malignancies. • An estimated 22,000 new cases occurred in the United States in 2012. • Almost all cases are thought to evolve from an asymptomatic premalignant stage termed monoclonal gammopathy of undetermined significance (MGUS). • The most common presenting symptoms are fatigue and bone pain. • Osteolytic bone lesions are the hallmark of the disease. • Hypercalcemia is found in one fourth of patients; the serum creatinine level is elevated in almost one half of patients. • Diagnosis requires 10% or more clonal plasma cells in the bone marrow and/or a biopsy-proven plasmacytoma, monoclonal (M) protein in the serum and/or urine (except in patients with true nonsecretory myeloma), and evidence of end organ damage (hypercalcemia, renal insufficiency, anemia, or bone lesions) attributable to the underlying plasma cell disorder. Patients with 60% or more clonal plasma cells in the bone marrow are considered to have multiple myeloma even in the absence of end organ damage. • M proteins can be detected by serum protein electrophoresis (SPEP) and immunofixation in 93% of patients; addition of urine protein electrophoresis (UPEP) and urine immunofixation or the serum free light chain (FLC) assay will increase sensitivity to 97% or higher. • The International Staging System (ISS) divides patients into three distinct stages and prognostic groups based on the β2-microglobulin and albumin levels in the serum. • High-risk myeloma is defined as the presence of any one or more of the following: deletion 17p, immunoglobulin heavy-chain (IgH) translocations t(14;16) or t(14;20), plasma cell leukemia, increased lactate dehydrogenase level, and high-risk signature on gene expression profiling studies. The translocation t(4;14) is considered intermediate risk, and all others indicate standard risk. • Newly diagnosed patients are categorized as having standard-, intermediate-, and high-risk myeloma based on specific prognostic factors. • Initial therapy for patients with standard-risk disease is with regimens such as lenalidomide–low-dose dexamethasone (Rd) or bortezomib-cyclophosphamide-dexamethasone (VCD). A bortezomib-containing regimen is preferred as initial therapy for patients with intermediate- and high-risk myeloma. • After 4 months of initial therapy, patients eligible for transplantation can pursue early or delayed autologous stem cell transplantation (ASCT). If early ASCT is used, a second ASCT is considered in patients who do not achieve a very good partial response or better with the first ASCT. If transplant is delayed, patients will continue on the induction chemotherapy drugs at lower doses until plateau or progression occurs. • Options for relapsed disease include thalidomide, lenalidomide, bortezomib, alkylating agents, anthracyclines, and corticosteroids alone or in combination. Monoclonal Gammopathy of Undetermined Significance (MGUS) • MGUS is an asymptomatic, premalignant, clonal plasma cell proliferative disorder defined by the presence of a serum M protein level less than 3 g/dL, bone marrow plasma cells less than 10%, and absence of anemia, hypercalcemia, lytic bone lesions, or renal failure that can be attributed to the plasma cell proliferative disorder. • MGUS is found in approximately 3% of the general population 50 years of age and older. • Patients with three adverse risk factors, namely, an abnormal serum FLC ratio, non-immunoglobulin G (IgG) MGUS, and a high serum M protein level (≥15 g/L), have a risk of progression to multiple myeloma or related malignancy at 20 years of 58% (high-risk MGUS) compared with 37% in patients with any two of these risk factors present (high-intermediate risk MGUS), 21% with one risk factor present (low-intermediate risk MGUS), and 5% when none of the risk factors was present (low-risk MGUS). • The current standard of care for MGUS is observation alone, without therapy. Smoldering Multiple Myeloma (SMM) • SMM is defined by the presence of a serum IgG or IgA M protein greater than or equal to 3 g/dL and/or bone marrow plasma cells of 10% to 60% and absence of anemia, hypercalcemia, lytic bone lesions, or renal failure that can be attributed to plasma cell proliferative disorder. • The risk of progression to myeloma or related malignancy is much higher in SMM compared with MGUS, at 10% per year versus 1% per year, respectively. • The standard of care is observation alone until evidence of progression to myeloma. • Waldenström macroglobulinemia is a clonal IgM M protein–secreting lymphoid/plasma cell disorder that currently also includes the entity referred to previously as lymphoplasmacytic lymphoma. • Median survival is approximately 5 years. • There are four options for initial therapy: rituximab, purine nucleoside analogs, alkylators, and combination chemotherapy. Unfortunately, there are no randomized data to determine the best option; therapy is typically decided based on the age of the patient and the aggressiveness of the presentation. • Options listed for initial therapy can also be tried at the time of relapse. The same initial therapy can be tried again at relapse if there was an adequate interval between cessation of therapy and relapse. Other options for relapsed, refractory disease include stem cell transplantation, interferon-α, thalidomide, and bortezomib. • Plasmapheresis is indicated for the treatment of hyperviscosity syndrome. Systemic AL (Immunoglobulin Light Chain) Amyloidosis • Amyloid is a fibrillar proteinaceous material detected with Congo red staining based on a characteristic apple-green birefringence under polarized light. • It consists of rigid, linear, nonbranching fibrils, 7.5 to 10 nm in width, aggregated in a β-pleated sheet conformation. There are several distinct types of amyloidosis classified based on the protein composition of the amyloid material. • AL (immunoglobulin light chain) amyloidosis refers to the type of amyloidosis derived from the variable portion of a monoclonal light chain. It should be suspected when patients with the appropriate clinical syndrome (e.g., nephrotic syndrome, axonal neuropathy, restrictive cardiomyopathy) display evidence of a plasma cell proliferative disorder such as a serum or urine M protein. • Patients are offered ASCT if eligible. Patients not eligible for ASCT (as shown by poor performance status, major comorbidities, three or more organs involved, and advanced cardiac amyloidosis) are treated with melphalan plus high-dose dexamethasone or with a bortezomib-based regimen. • Thalidomide (or lenalidomide) plus dexamethasone are second-line treatment options for patients with systemic AL amyloidosis. • Solitary plasmacytomas may be confined to bone (solitary bone plasmacytoma) or occur in extramedullary sites (extramedullary plasmacytoma). • Patients with solitary plasmacytoma are at risk for progression to multiple myeloma. • Treatment consists of radiation in the range of 40 to 50 Gy to the involved site. • Disease-free survival rate at 10 years ranges from 25% to 50%. The diagnosis of active myeloma requires 10% or more plasma cells on bone marrow examination and/or biopsy-proven plasmacytoma, monoclonal (M) protein in the serum and/or urine (except in patients with true nonsecretory myeloma), and evidence of end organ damage (hypercalcemia, renal insufficiency, anemia, or bone lesions) that is attributable to the underlying plasma cell disorder (Table 104-1).3–3 Patients with 60% or more clonal plasma cells in the bone marrow are also considered to have multiple myeloma even in the absence of end organ damage.4 Table 104-1 Diagnostic Criteria for Plasma Cell Disorders POEMS, Polyneuropathy, organomegaly, endocrinopathy, monoclonal protein, skin changes. *The source data do not define an optimal cutoff value for considering elevated VEGF level as a major criterion. We suggest that VEGF measured in the serum or plasma should be at least threefold to fourfold higher than the normal reference range for the laboratory that is doing the testing to be considered a major criterion. †To consider endocrinopathy as a minor criterion, an endocrine disorder other than diabetes or hypothyroidism is required, because these two disorders are common in the general population. Modified from Kyle RA, Rajkumar SV. Criteria for diagnosis, staging, risk stratification and response assessment of multiple myeloma. Leukemia 2009;23:3–9. Multiple myeloma accounts for approximately 10% of hematologic malignancies.5,6 The annual incidence, age-adjusted to the 2000 U.S. population, is 4.3 per 100,000.7 An estimated 22,000 new cases and 11,000 new deaths were expected to occur in the United States in 2012.8 Multiple myeloma is twice as common in blacks compared with whites and slightly more common in males than females.9 The median age at diagnosis is 66 years,10 and only 2% of patients are younger than 40 years of age. Almost all patients with myeloma are believed to evolve from an asymptomatic premalignant stage termed monoclonal gammopathy of undetermined significance (MGUS).11,12 MGUS is present in more than 3% of the population older than the age of 50 and progresses to myeloma or related malignancy at a rate of 1% per year.13,14 In some patients, an intermediate asymptomatic but more advanced premalignant stage referred to as smoldering multiple myeloma (SMM) can be recognized clinically.15 MGUS is characterized by evidence of genomic instability on molecular genetic testing. The trigger for this genomic instability is not well understood, but current evidence suggests that antigenic stimulation may be a key factor (Fig. 104-1). Unlike normal plasma cells, human myeloma cell lines and primary myeloma cells express a broad range of Toll-like receptors (TLRs). TLRs are normally expressed by B lymphocytes and are essential for these cells to recognize infectious agents and pathogen-associated molecular patterns (PAMP), which then initiates the host-defense response.18–18 The aberrant expression of TLRs by plasma cells may enable them to respond to TLR-specific ligands, resulting in an abnormal and perhaps sustained response to infection. It has been shown that TLR-specific ligands cause increased myeloma cell proliferation, survival, and resistance to dexamethasone-induced apoptosis. These effects are mediated in part by autocrine interleukin (IL)-6 production.16,17 IL-6 is a major growth factor for plasma cells,19 and there is overexpression of CD126 (IL-6 receptor α-chain) in MGUS compared with normal plasma cells.20,21 Thus, abnormal TLR expression and/or overexpression of IL-6 receptors in plasma cells may be early, initiating events that lead to an abnormal response to infection and act as sustained, autocrine IL-6–dependent, proliferative triggers for plasma cells. During this process, plasma cells may acquire one of various cytogenetic alterations that result in a limited clonal plasma cell proliferative process, namely MGUS. Immunosuppression either by promoting evasion of tumor surveillance or by promoting antigenic stimulation may also contribute to the initiation of monoclonal gammopathies. Monoclonal proteins have been reported in the context of immunosuppressive states such as bone marrow/stem cell transplantation, organ transplantation, and human immunodeficiency virus (HIV) infection.22–25 Patients undergoing renal transplantation develop M proteins depending on the level of immunosuppression to which they are subjected.25 Over 90% of MGUS is associated with cytogenetic changes possibly precipitated by infection or immune dysregulation as discussed earlier, likely during IgH switch recombination or somatic hypermutation.26 Approximately 50% of patients with MGUS have primary translocations in the clonal plasma cells involving the immunoglobulin heavy chain (IgH) locus on chromosome 14q32 (IgH-translocated MGUS/SMM) (see Fig. 104-1).5,26 The most common partner chromosome loci and genes dysregulated in these translocations are 11q13 (CCND1 [cyclin D1 gene]), 4p16.3 (FGFR3 and WHSC1), 6p21 (CCND3 [cyclin D3 gene]), 16q23 (MAF), and 20q11 (MAFB).29–29 It is likely that these translocations play an important pathogenetic role in the resultant limited clonal proliferation that is clinically manifested as MGUS. Approximately 45% of cases of MGUS are associated with trisomies, usually of the odd numbered chromosomes with the exception of 13; and the origin of the remaining 5% or fewer of MGUS is not clear.26,30 These categories of MGUS that lack evidence of IgH translocations are referred to as non–IgH-translocated MGUS. In one recent study it was shown that there is a small overlap wherein a subset of patients has both types of translocations as well as trisomies; this condition is likely present at the MGUS stage, but the sequence and prevalence are not well known (Table 104-2).31 Table 104-2 Revised Primary Molecular Cytogenetic Classification of Myeloma *In addition to the effect of the translocated gene, there is the added effect of trisomies of odd-numbered chromosomes in this group. Modified from Kumar S, Fonseca R, Ketterling RP, et al. Trisomies in multiple myeloma: impact on survival in patients with high-risk cytogenetics. Blood 2012;119:2100 American Society of Hematology. Deletion of chromosome 13, a major prognostic factor in multiple myeloma, is seen in up to 50% of patients with MGUS by interphase fluorescent in-situ hybridization (FISH); hence, the presence of this abnormality cannot be used to differentiate MGUS from multiple myeloma.29 The constant rate of progression of MGUS to myeloma, macroglobulinemia, or related malignancy in a recent epidemiological study over a period spanning 30 to 35 years strongly suggests a simple, random, two-hit genetic model of malignancy.13 The risk of progression is similar regardless of the known duration of antecedent MGUS, suggesting that the second-hit responsible for progression is a random event, not cumulative damage. The specific second-hit that initiates the cascade of events associated with progression is unknown. Several abnormalities have been detected with progression in both the plasma cell and its microenvironment that likely play a role in the progression of MGUS, but little is known about the sequence of events (see Fig. 104-1). RAS mutations, CDKN2A methylation, abnormalities involving the MYC family of oncogenes, secondary translocations, and TP53 mutations have all been identified in clonal plasma cells in association with progression to the symptomatic stage.27 It is possible that the second-hit responsible for disease progression may be different in IgH-translocated versus non–IgH-translocated MGUS, and even within the various subtypes of IgH-translocated MGUS, depending on the partner chromosome involved. For example, amplification of chromosome 1q21 has been noted in over 40% of patients with SMM and myeloma compared with 0% in MGUS,32 suggesting that such amplification (e.g., by trisomy 1) may play a role in progression, perhaps in non–IgH-translocated MGUS. The bone marrow microenvironment undergoes marked changes with progression, including induction of angiogenesis,33 suppression of cell-mediated immunity,34 and paracrine loops involving cytokines such as interleukin-6 and vascular endothelial growth factor (VEGF).35 As in solid tumors, the transition from MGUS to multiple myeloma may involve an angiogenic switch. In solitary plasmacytoma, which can be considered to be analogous to localized stage I solid tumor, induction of angiogenesis at the time of diagnosis has been shown to be a predictor of progression to myeloma, suggesting a pathogenetic role for the process in disease progression.36 Furthermore, there is a gradual increase in degree of bone marrow angiogenesis along the disease spectrum from MGUS to SMM to symptomatic myeloma.33 In one study, approximately 60% of myeloma bone marrow plasma samples stimulated angiogenesis in an in vitro angiogenesis assay, compared with 0% of SMM and 7% of MGUS (P < .001).37 There are several important mechanisms that mediate increased osteoclast activation.38 There is an increase in the receptor activator of nuclear factor κB (NF-κB) ligand (RANKL) expression by osteoblasts and possibly plasma cells. This is accompanied by decreased stromal cell secretion of the RANKL decoy receptor osteoprotegerin (OPG).39,40 In addition, the binding of OPG to RANKL is also inhibited by syndecan-1 (CD138) secreted and or shed by myeloma cells. The net result of these facts is an increase in the RANKL/OPG ratio. This causes increased osteoclast activation mediated through the NF-κB pathway. A second factor governing the activation of osteoclasts is the release of macrophage inflammatory protein (MIP)-1α and MIP-1β by myeloma cells. Both these cytokines cause osteoclast activation primarily by increasing RANKL expression in stromal cells. Finally, there is increased expression of stromal derived factor-1α (SDF-1α) by stromal cells and myeloma cells. SDF-1α causes osteoclast activation by binding to chemokine (C-X-C motif) receptor 4 (CXCR4) on osteoclast precursors. In addition to these three factors, several other cytokines such as IL-1β and IL-6 are also thought to play a role in osteoclast activation and bone resorption. Osteoblast inhibition in myeloma is believed to be primarily related to increased dickkopf 1 (DKK1) expression by myeloma cells.41 DKK1 binds to Wnt receptors and inhibits the Wnt signaling pathway by preventing the normal binding of Wnt glycoproteins to Wnt receptors.38 The inhibition of Wnt signaling prevents the intracellular stabilization of β-catenin, leading to the phosphorylation and degradation of β-catenin through the proteasome pathway. Normally, β-catenin plays an important role in osteoblast activation and its absence reduces the activity of osteoblasts. In addition to its key role in causing osteoblast inhibition, increased levels of DKK1 may also contribute in some measure to osteoclast activation. Other factors such as IL-3 and IL-7 may contribute to osteoblast inhibition. The combination of osteoclast activation and inhibition of osteoblast differentiation is believed to be the mechanism behind the development of osteolytic lesions in myeloma. The most common presenting symptoms of myeloma are fatigue and bone pain.10 Osteolytic bone lesions and/or compression fractures are the hallmark of the disease and can be detected on routine radiography, magnetic resonance imaging (MRI), computed tomography (CT), or combined fluorodeoxyglucose-labeled positron emission tomography (FDG-PET)/CT (Figs. 104-2 through 104-4). Bone pain may present as an area of persistent pain or be migratory, often in the lower back and pelvis. Pain may be sudden in onset when associated with a pathological fracture and is often precipitated by movement. Extramedullary expansion of bone lesions may cause nerve root or spinal cord compression. Anemia occurs in 70% of patients at diagnosis and is the primary cause of fatigue. Hypercalcemia is found in one fourth of patients, whereas the serum creatinine value is elevated in almost one half of patients. M proteins can be detected by serum protein electrophoresis (SPEP) in 82% of patients with myeloma and by serum immunofixation (IFE) in 93% of patients.10 Up to 20% of patients with myeloma lack heavy-chain expression in the M protein and are considered to have light-chain myeloma. The M protein in these patients is always detected in the urine but can be absent in the serum even by immunofixation, making it imperative that protein electrophoresis and immunofixation are always done on both the serum and urine in all patients in whom myeloma is suspected. Addition of urine protein electrophoresis (UPEP) and urine IFE will increase the sensitivity of detecting M proteins in patients with myeloma to 97%. Most (60%) of the remaining patients who are negative for M proteins on serum and urine electrophoresis and immunofixation studies will have evidence of clonal paraproteins on the serum free light chain (FLC) assay. Currently, only 1% to 2% of patients with myeloma will have no detectable M proteins on any of these tests. These patients have true nonsecretory myeloma. Agarose gel SPEP and immunofixation are the preferred methods of detection of serum M proteins (Fig. 104-5). M proteins appear as a localized band on SPEP.42 After recognition of a localized band suggestive of an M protein on SPEP, immunofixation is necessary for confirmation and to determine the heavy- and light-chain class of the M protein. In addition, immunofixation is more sensitive than SPEP and allows detection of smaller amounts of M protein and should therefore be performed whenever myeloma, amyloidosis, macroglobulinemia, or a related disorder is suggested. The size of the M protein is measured using SPEP. For purposes of clinical trials and monitoring, the M protein is considered to be “measurable” if it is 1 g/dL or more in the serum and or greater than or equal to 200 mg/d in the urine. The serum FLC assay (Freelite, The Binding Site Limited, Birmingham, UK) provides an important tool to quantify monoclonal light chains secreted by myeloma cells, especially in patients who secrete small amounts of intact monoclonal immunoglobulin.43 This automated nephelometric assay allows quantitation of free kappa (κ) and lambda (λ) chains (i.e., light chains that are not bound to intact immunoglobulin) secreted by plasma cells. An abnormal κ/λ FLC ratio indicates an excess of one light chain type versus the other and is interpreted as a surrogate for clonal expansion based on extensive testing in normal volunteers and patients with myeloma, amyloidosis, and renal dysfunction.43,44 The normal serum free κ level is 3.3 to 19.4 mg/L, and the normal free λ level is 5.7 to 26.3 mg/L.45 The normal ratio for FLC κ/λ is 0.26 to 1.65. The normal reference range in the FLC assay reflects a higher serum level of free λ light chains than would be expected given the usual κ/λ ratio of 2 for intact immunoglobulins. This occurs because the renal excretion of free κ (which exists usually in a monomeric state) is much faster than that of free λ (which is usually in a dimeric state). 43,44 Patients with a κ/λ FLC ratio less than 0.26 are typically defined as having monoclonal λ free light chain and those with ratios greater than 1.65 are defined as having a monoclonal κ free light chain. If the FLC ratio is greater than 1.65, κ is considered to be the “involved” FLC and λ the “uninvolved” FLC, and vice versa if the ratio is less than 0.26. A study of 428 patients demonstrated that the urine studies can be eliminated when screening for the presence of monoclonal plasma cell disorders by using the serum FLC assay in combination with SPEP and immunofixation.46 If a monoclonal plasma cell disorder is identified on screening, urine studies should be done to aid monitoring disease progression and response to therapy over time. Besides its role as a substitute for urine studies in the screening of plasma cell disorders, the FLC assay is used to predict prognosis in MGUS, SMM, and solitary plasmacytoma.49–49 It is also used to monitor patients with oligosecretory or nonsecretory myeloma, primary amyloidosis, and patients with the light-chain only form of myeloma.44,50–52 To use the FLC assay to monitor disease progression, the baseline FLC ratio must be abnormal and the involved FLC level 100 mg/L or higher.52 Plain radiographic examination of all bones including long bones (skeletal survey) is the preferred method of detecting lytic bone lesions in myeloma. Conventional radiographs show skeletal abnormalities in almost 80% of patients with myeloma; often these lesions have a characteristic punched-out appearance. Osteoporosis and/or fractures are also detected by conventional radiography. Occasionally osteosclerotic lesions can occur. CT and MRI are more sensitive than conventional radiography in detecting bone disease. Among asymptomatic multiple myeloma patients with normal radiographs, up to 50% have tumor-related abnormalities on MRI of the lower spine. FDG-PET/CT is are useful in the evaluation of bone disease, in staging, and in assessing response to therapy in myeloma (Fig. 104-6).53 It is not necessary in all patients with myeloma but is of value in patients in whom there is uncertainty about the extent of bone disease. It is also of value in monitoring response to therapy in patients with oligosecretory myeloma. In most cases, if there is uncertainty about the extent of disease, FDG-PET/CT is preferable to MRI or CT because the whole body can be imaged. However, if more detailed imaging is required, as in the case of spinal cord compression, MRI or detailed CT may be preferable. The role of bone mineral density studies in myeloma and the use of these studies in identifying patients at risk for pathological fractures and prophylactic bisphosphonate therapy remain unresolved. However, if these studies have been done, the results can be used to guide the frequency of bisphosphonate administration. Given their impact on prognosis (see later discussion), bone marrow samples should be studied with conventional karyotyping studies and/or FISH to detect specific abnormalities such as t(4;14), t(14;16), and 17p−. The bone marrow plasma cell proliferative rate should be estimated by multiparametric flow cytometry, if available. Gene expression profiling studies, if done, can provide additional prognostic information.54,55
Multiple Myeloma and Related Disorders
Multiple Myeloma
Introduction
Definition
Epidemiology
Pathogenesis
Transition from Normal Plasma Cell to MGUS
Antigenic Stimulation and Immunosuppression
Cytogenetic Changes
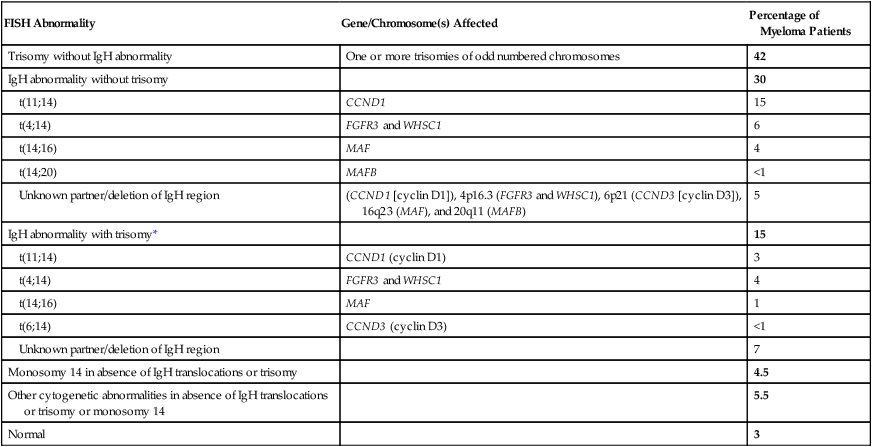
FISH Abnormality
Gene/Chromosome(s) Affected
Percentage of Myeloma Patients
Trisomy without IgH abnormality
One or more trisomies of odd numbered chromosomes
42
IgH abnormality without trisomy
30
t(11;14)
CCND1
15
t(4;14)
FGFR3 and WHSC1
6
t(14;16)
MAF
4
t(14;20)
MAFB
<1
Unknown partner/deletion of IgH region
(CCND1 [cyclin D1]), 4p16.3 (FGFR3 and WHSC1), 6p21 (CCND3 [cyclin D3]), 16q23 (MAF), and 20q11 (MAFB)
5
IgH abnormality with trisomy*
15
t(11;14)
CCND1 (cyclin D1)
3
t(4;14)
FGFR3 and WHSC1
4
t(14;16)
MAF
1
t(6;14)
CCND3 (cyclin D3)
<1
Unknown partner/deletion of IgH region
7
Monosomy 14 in absence of IgH translocations or trisomy
4.5
Other cytogenetic abnormalities in absence of IgH translocations or trisomy or monosomy 14
5.5
Normal
3
Progression of MGUS to Malignancy
Pathogenesis of Bone Lesions
Clinical Presentation
Investigation
Identification of Monoclonal Proteins
Serum Protein Electrophoresis and Immunofixation
Serum Free Light-Chain Assay
Identification of Bone Disease
Bone Marrow Studies
Multiple Myeloma and Related Disorders