ATA risk level
RET mutation codon(s)
MEN2 subtype
Prophylactic thyroidectomy
Screening for PHEO
Screening for PHPT
A
649, 768, 790, 791, 804, 891
FMTC
When calcitonin rises/5–10 years age
Start at 20 years age, then periodically
Start at 20 years age, then periodically
B
609, 611, 618, 620, 630, 631
FMTC and MEN2A
Before 5 years age
Start at 20 years age, then annually
Start at 20 years age, then annually
C
634
MEN2A
Before 5 years age
Start at 8 years age, then annually
Start at 8 years age, then annually
D
883, 918
MEN2B
At diagnosis
Start at 8 years age, then annually
Not needed
Based on the recommendations from the American Thyroid Association [15] and the International Workshop on Multiple Endocrine Neoplasia [11], a risk-based stratification has been developed for MEN2 patients [1]. As seen Table 31.1, Group D patients with mutations in codon 918 and 883 are at the highest risk, and for this reason prophylactic thyroidectomy is recommended within the first months of life and screening for pheochromocytoma should start at age 8 years and continue annually. In contrast, patients in group A present with MTC an at older age and prophylactic thyroidectomy can be performed later in life. In addition, for group A patients screening for pheochromocytoma and PTH should begin in adolescence.
Genetic Screening
Historically, the diagnosis of MTC in patients with MEN2A or MEN2B depended on the demonstration of elevated plasma calcitonin levels either at baseline or after intravenous administration of the potent secretagogues calcium or pentagastrin. Advances in the molecular genetics underlying the MEN2 syndromes have resulted in DNA testing becoming the optimal test for their detection. Currently, genetic analysis identifies more than 95% of responsible mutations [21].
DNA testing for MEN2 is based on identification of RET mutations, and is considered the standard of care for newly identified MTC patients, regardless of age at diagnosis or family history[10]. Even in children with apparent sporadic MTC all exons should be sequenced and if no mutation is found, tumor DNA can be analyzed. Early diagnosis by screening of family members at risk in MEN2 kindreds is essential because testing can identify family members who are at risk for MTC and this life-threatening disease can be cured or prevented by early thyroidectomy. Moreover, knowledge of genotype–phenotype correlations may help estimate the chances of developing additional endocrine tumors, provide prognostic information, and plan surgical management.
Genetic testing can be useful in 3 groups of patients. First, in patients with histologically proven MTC or C-cell hyperplasia it can help differentiate between sporadic and hereditary MTC. If patients have hereditary MTC then their families should be screened. Next, in patients with familial occurrence of MTC and/or pheochromocytoma, genetic testing can differentiate between the subtypes of MEN2, identify the risk of developing other tumors, and determine when to start their screening. More importantly, genetic testing in MEN2 patients guides the timing for prophylactic thyroidectomy [11, 22]. Lastly, in first degree family members of MEN2 patients genetic testing can distinguish gene carriers from non carriers. Documentation that the RET mutation is not present eliminates the need for ongoing screening for MEN2 tumors and avoids unnecessary thyroidectomy in genetically normal subjects in MEN2 families. It is known from retrospective DNA testing, that such subjects can have falsely positive calcitonin level with pentagastrin stimulation that would suggest they have MEN2 [11].
Diagnosis and Management
Medullary Thyroid Cancer
Approximately 5% of thyroid neoplasms in children are medullary carcinomas, arising from the parafollicular C cells. MTC is usually the first tumor to develop in MEN2 patients and is the most common cause of death in this group. The malignant transformation of C cells starts early in life and C-cell hyperplasia and micro-MTC can be found as early as 2 months in some kindreds. This is especially true in MEN2B, which typically has a more aggressive form of MTC [23]. As with other thyroid neoplasms, the clinical diagnosis of MTC is often made with palpation of a thyroid nodule or after there is significant spread of the tumor to the adjacent cervical lymph nodes or to distant sites (lung, liver and bone) [24]. Also, diarrhea can be seen when there is significant hypercalcitoninemia and is usually a poor prognostic sign [25]. Less frequently, the tumor can be recognized after the diagnosis of pheochromocytoma or hyperparathyroidism. Rarely, MTC has been shown to cause Cushing’s syndrome due to ectopic production of corticotropin (ACTH).
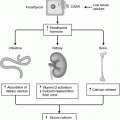
Calcitonin production is increased in C-cell hyperplasia and MTC, and measurements of plasma calcitonin are used as a tumor marker to monitor the presence and progression of MTC. Usually, serum calcitonin levels correlate with tumor mass and tend to be high (>1000 pg/m) in patients with a palpable tumor on exam. Likewise, elevated plasma calcitonin levels after tumor resection suggests persistent or recurrent disease [1].
The only effective treatment for MTC is surgical resection, underscoring the importance of early diagnosis and therapy before metastasis occurs. For this reason, current management of MTC in children from families having the MEN2 syndrome relies on the presymptomatic detection of the RET proto-oncogene mutation responsible for the disease. Affected children with MEN2A should undergo total thyroidectomy at about the age of 5 years, before the cancer spreads beyond the thyroid gland [26]. Indeed, approximately 80% of children who have thyroidectomy based on the presence of the RET mutation will already have foci of MTC within the thyroid gland [27]. As a matter of fact, almost all prophylactic thyroidectomies performed in recent years have shown that various stages of C-cell disease are present between 1 and 6 yr of age in patients of MEN2 families. Owing to the increased virulence of the MTC in children having MEN2B, it may be preferable for their thyroid glands to be removed in infancy. As discussed before, the specific RET mutation can help determine the timing of prophylactic thyroidectomy. Patients with MTC should be evaluated for possible pheochromocytoma before thyroidectomy and if the diagnosis is made, adrenalectomy should be performed first.
Because of the high incidence of bilateral disease in MTC, complete removal of the thyroid gland (total thyroidectomy) is the recommended for surgical management of MTC in children [28], as well as, for carriers of the mutation. The extent of neck lymph node dissection and the management of devascularized parathyroid glands differ depending upon the patient’s MEN2 subtype, patient symptoms and whether the intervention is prophylactic or therapeutic. Also, the preoperative imaging may help determine how extensive the lymph node dissection should be. Usually, the dissection includes lymph nodes in the central compartment of the neck, medial to the carotid sheaths and between the hyoid bone and the sternum. This central lymph node dissection may be unnecessary in patients with MEN2A under the age of 11[29].
Single or combination chemotherapy has been ineffective in the treatment of unresectable or metastatic MTC. The antiangiogenesis and tyrosine kinase inhibitor drugs, such as combretastatin and imatinib mesylate, have been observed to have antineoplastic effects on MTC in vitro. Clinical trials need to be performed to determine the usefulness of these agents [21, 30–32]. Recent trials with VELF inhibitors have shown some promise in arresting MTC progression in patients with metastatic disease [33].
Palliative radiation therapy can also be used to diminish the tumor burden in the neck and to prevent local recurrence. Whether morbidity or mortality is improved remains unclear [21]. High-dose 131iodine anticarcinoembryonic antigen antibody treatment combined with post-therapy autologous hematopoietic stem cell rescue has limited toxicity, but the antitumor effect is unknown [34].
The postoperative management and monitoring of patients with MTC in the setting of MEN2 includes periodic physical exams, serial monitoring of serum calcitonin concentrations and neck ultrasound if the calcitonin is elevated. Thyroid hormone replacement is used to maintain euthyroidism and calcium supplementation is given when needed. After adolescence, yearly biochemical screening for pheochromocytoma is recommended.
Pheochromocytoma
Pheochromocytomas are rare catecholamine-secreting tumors of the adrenal medulla. It affects 40% of patients with MEN2A and MEN2B, although there is large variability of its penetrance among kindreds. In patients with MEN2 it is rare for a pheochromocytoma to be the initial manifestation and present before the development of MTC, but it has been reported [35]. Usually, pheochromocytomas become evident about 10 years after the diagnosis of C-cell hyperplasia or MTC [36]. As with MTC, the penetrance of pheochromocytoma among different MEN 2 kindreds depends on specific RET germline mutations [37].
In general, about 10% of children with pheochromocytoma have familial disease [38, 39]. Because these familial cases may include MEN2 with the risk of MCT, any child who presents with a pheochromocytoma should be screened for MEN2. Pheochromocytomas are found in an extra-adrenal location in 30% of children [39], but this is rare in MEN2 [37].
The signs and symptoms associated with pheochromocytomas in patients with MEN2 include paroxysmal hypertension which can be extreme and episodic. Also, orthostatic hypotension, when present, is suggestive of MEN2. Occasionally, patients present with catecholamine-induced cardiomyopathy and hypertensive encephalopathy [40]. Laboratory confirmation is similar to sporadic pheochromocytomas, specifically the finding of increased plasma metanephrine [41]. Once there is biochemical confirmation of pheochromocytoma the tumor should be localized and possibility of, bilateral disease evaluated using MRI.
Pharmacologic blockade of excess catecholamines is a critical part of preoperative preparation of the patient with pheochromocytoma. The goals are to replete the intravascular volume, treat severe hypertension, control cardiac arrhythmias if present, and minimize the risk of perioperative cardiovascular instability.
The definite treatment for pheochromocytoma remains surgical removal of the lesion. Pheochromocytomas can be extraordinarily active for their size, and accurate preoperative localization of the lesions is essential. In the setting of bilateral disease, partial adrenalectomy should be considered [42]. In a series reported by Yip et al. [43], 65% of patients did not require chronic corticosteroid replacement after partial adrenalectomy in marked contrast to those who underwent total bilateral adrenalectomy. They also showed that the risk of recurrent pheochromocytoma in the adrenal gland remnant after the cortical-sparing procedure was about 10%.
Surgical management of unilateral disease is a topic of controversy. Some advocate bilateral adrenalectomy because about 30% of patients undergoing unilateral surgery may eventually develop pheochromocytoma in the remaining adrenal gland and would require resection [44]. Nevertheless, this may not happen for many years, and initial unilateral adrenalectomy would avoid the need for steroid replacement in the interval. Also favoring avoidance of initial bilateral adrenalectomy is the observation that no lethal complications from catecholamine crisis and no metastatic disease occurred in patients with MEN2 who underwent unilateral adrenalectomy and then developed contralateral disease [44].
Laparoscopic adrenalectomy is now the preferred approach for benign functioning adrenal tumors, including pheochromocytoma [45, 46]. Normalization of the blood pressure should be expected if all functioning pheochromocytoma tissue is removed. Children who have had resection of a pheochromocytoma should undergo examination with blood pressure measurements and urine catecholamine levels twice per year to monitor for recurrence. Patients with familial pheochromocytomas should be followed especially carefully for the development of disease in the opposite gland [47, 48]. For additional discussion of pheochromocytoma see Chap. 8.
Primary Hyperparathyroidism
PHPT in children is extremely rare with an incidence of 2–5 per 100,000, with most cases (80%) occurring during adolescence [49, 50]. In large series of patients with MEN2 clinically evident hyperparathyroidism was noted at a median age of 38 years (ranging from 7 to 71 years) [51–53] but it has been described in a patient as young as 5 years of age, who was found to have a pathologic parathyroid gland at time of his prophylactic total thyroidectomy [54]. In most MEN2 patients (75–80%) PHPT is diagnosed at the same time as MTC or pheochromocytoma and the rest are found at follow up after thyroidectomy [11]. Less than 5% of cases PHPT in MEN2 patients are diagnosed before the presentation of MTC or pheochromocytoma [53].
Different from the usual sporadic cases, PHPT in patients with MEN 2A is characterized by multigland hyperplasia and an increased incidence of ectopic or supernumerary glands. Signs and symptoms of hypercalcemia in PHPT tend to be more severe in children than in adults and can be associated with end-organ damage, making early diagnosis critical.
Parathyroid disease seems to develop more rarely in patients with MEN2A who undergo a total thyroidectomy at an early age [55, 56]. Some theories attribute this phenomenon to removal of normal parathyroid glands during thyroidectomy. This could decrease the incidence of parathyroid disease in two ways—either by reducing the number of parathyroid glands at risk, or by removing the stimulus for parathyroid growth after removal of C cells [55, 57].
The diagnosis of primary hyperparathyroidism is confirmed with the findings of hypercalcemia and inappropriately high-serum parathyroid hormone (PTH) concentrations. In a patient with known or presumed MEN2A, the indications for surgical intervention are similar to those in patients with sporadic primary hyperparathyroidism [58].
Although the frequency of PHPT in MEN 2A is not high, and the symptoms of hypercalcemia tend to be milder, the surgical treatment options are similar to those for patients with MEN1. Most of these patients have multiple parathyroid tumors, so bilateral exploration of previously nonoperative patients should be planned regardless of the outcome of preoperative localization studies. Such imaging studies may nonetheless be anatomically helpful to the surgeon and can therefore be indicated. These studies (ultrasonography, sestamibi imaging, or chest CT) are certainly recommended before re-operation in patients with recurrent or persistent disease.
There is some disagreement regarding the preferred surgical management with arguments and challenges similar to those for patients with hyperparathyroidism in the setting of MEN1 [52, 59, 60]. Because symptoms are milder and the likelihood of recurrent HPT is lower than in MEN1, Dotzenrath [59] and O’Riordain [60] both recommend a conservative approach with excision of only enlarged glands without transcervical thymectomy. This technique is associated with low rate of persistent and recurrent PHPT, inferring that it offers a better chance to avoid postoperative hypoparathyroidism [56]. On the other hand, it may be wise to concomitantly undertake cervical thymectomy at the initial surgery especially in patients with multiple gland involvement. For patients with more severe hypercalcemia, however, a more aggressive approach may be employed. Some surgeons recommend three and one-half-gland parathyroidectomy, (or all but one-half gland, if supernumerary glands are found), together with thymectomy, and with subtotal parathyroidectomy being repeated if the patient has a recurrence [60, 61]. On the other hand, others prefer to remove all the glands and heterotopically transplant some tissue into the nondominant forearm [62]. The latter approach has the advantage of avoiding repeated neck exploration if hyperparathyroidism should recur, and has been shown to be safe in infants and children [27, 63, 64]. Moreover, total parathyroidectomy with heterotopic autotransplantation has been shown to result in improved survival in infants with severe hypercalcemia [62]. As noted previously, patients with total parathyroidectomy and autotransplantation require a short period of vitamin D and calcium supplementation until the heterotopic tissue begins to function. If, however, the autograft does not function the patient may have permanent hypoparathyroidism.
Overall cure rates can be expected to be as high as 95%. Patients with MEN2 should have annual screening for hyperparathyroidism by serum calcium and intact parathyroid hormone level measurements.
Hirschsprung’s Disease
RET mutations are responsible for up to 50% of familial cases of Hirschsprung’s Disease (HD) and may also be seen in sporadic HD cases [65]. Interestingly, the clinical association between aganglionic megacolon, megaloureter, pheochromocytoma, and neuromatosis [66] was actually reported before the first MEN descriptions [65].
There are multiple publications noting the correlation of MEN2A and HD. HD was found in 57% of children in families with a C620 mutation in exon 10 with a low penetrance [67–69]. The later, as well as mutations in C618 position are the most likely to predispose to HD [69]. Even though, MEN2A and HD arise from the same gene, they act through different pathways to activate RET kinase. They arise from variations on different parts of the gene, and have both extra and intracellular targets of protein transformation [70].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


