The association between thyroid carcinoma and pheochromocytoma (PHEO) was originally reported by Sipple, and in 1968 named as the multiple endocrine neoplasia type 2 syndrome (MEN 2) associated with medullary thyroid carcinoma (MTC).1–4 In 1975, MEN 2B was recognized with a previously reported characteristic phenotype.5,6
MEN 2 is an autosomal, dominant hereditary syndrome, with a 50% risk for offsprings of a carrier to inherit the syndrome. MEN 2 is present in 25% of MTC patients with prevalence of one per 30, 000 individuals.7–12 The MEN 2 genetic disorder includes MEN 2A, familial medullary thyroid carcinoma (FMTC), and MEN 2B. The MEN 2 gene was in 1987 located to chromosome 10q11.2, and in 1993/1994 the varieties of the syndrome, MEN 2A, FMTC, and MEN 2B, were demonstrated to be due to germline missense mutations in the RET (REarranged during Transfection) protooncogene.7–19 The germline RET gene mutations results in expression of abnormally overactive Ret protein in tissues where it is expressed and leads to the specific hereditary syndromes.7 Somatic RET mutations that occur later in life and are limited to C cells are present in 40% to 50% of sporadic MTCs. Founding de novo mutations arise exclusively from the paternal allele in MEN 2B, and in 10% of MEN 2A cases.20,21 Somatic RET mutations have been present in 40% to 50% of sporadic MTC, and in presence of codon 918 mutations been associated with aggressive tumors.22,23 Somatic RET mutations also occur in sporadic PHEO. Somatic RET rearrangements are seen in papillary thyroid carcinoma in children, who have been offers of radiation exposure.
The RET protooncogene with 21 exons encodes a transmembrane tyrosine kinase receptor (Fig. 48-1). The receptor consists of a transmembrane domain, an extracellular domain with a ligand-binding site, and an intracellular tyrosine kinase (TK) domain, and is expressed mainly in neuronal and neuroepithelial cells of neural crest origin.17,24 Mutation in the intracellular catalytic core (mutation of codon 918) has the highest transforming activity, whereas interference with ATP binding (mutations in codons 768, 790, 791, 804, and 891) has low transforming capacity.17,24 Germline “gain-of-function” mutations activate the RET receptor kinase causing thyroid C-cell hyperplasia (CCH), adrenomedullary hyperplasia, and parathyroid hyperplasia.17,24 Age-related progression from CCH to MTC, and development of PHEO, and primary hyperparathyroidism (PHPT) correlate with the transforming capacity of the different RET mutations, but require additional somatic second hits.7,24 MTC is generally the first neoplastic manifestation because of high susceptibility of C cells to the oncogenic RET activation.7,11
RET gene testing should be done prior to surgery in all patients with diagnosed MTC, since 10% of apparently sporadic MTC have had mutations. Gene testing detects approximately 98% of gene carriers, and will help with risk assessments for family members, and reveal the need for α-blockade if PHEO should be present. Routine RET gene testing is recommended also in patients with diagnosed PHEO. Only exons 10, 11, 13, 14, 15, and 16 need to be routinely tested; if initial sequencing is negative, special laboratories may sequence also exon 8 or the entire coding region.7
Medullary thyroid carcinoma (MTC) originates in calcitonin producing thyroid parafollicular C cells, occurring most frequently in a dorsal parts of the upper and middle thyroid lobes.7,10 Nearly 10% of patients with apparently sporadic MTC have been found to harbor germline RET gene mutations.12,25 Hereditary MTC occurs at younger age than sporadic MTC, is associated with CCH, and is frequently multicentric, and bilateral in contrast to solitary, sporadic MTC. Tumor cells have granulated cytoplasm, stain for calcitonin, and amyloid (with Congo red reactive with the calcitonin prohormone). CCH consists of C-cell clusters dispersed in-between the thyroid follicles, and is a preneoplastic condition in patients with RET mutations, which may progress to microscopic invasive MTC with variable time required for progression.7,10 There is a graded progression from a cellular to more fibrous growth patterns as the MTC ages.7 Serum calcitonin values serve as tumor marker of MTC. CCH and slightly raised serum calcitonin values occur also in 20% to 30% of normal individuals associated with aging, renal failure, HPT, hypergastrinemia, and chronic lymphocytic thyroiditis.7,26 Lymph node metastases from MTC relate to tumor size, being 20% to 30% for tumors <1 cm, 50% for tumors measuring 1 to 4 cm, and up to 90% for tumors >4 cm.27 Distant metastases occur to liver, bone, and lung. Liver metastases are often initially small (1 to 3 mm), and easily missed by computed tomography (CT), magnetic resonance imaging (MRI), or contrast-enhanced ultrasound (US) imaging.18 F-DOPA-PET, 5-HTP -PET, and FDG-PET combined with 3-phase vascular enhanced CT have been used with 50% to 80% sensitivity for detection of metastases.28,29 Since metastases may be small and with miliary spread, laparoscopy has sometimes been used to safely exclude liver metastases prior to extensive neck reoperations. Around 50% of index patients with MEN 2 present with palpable, and locally advanced disease, with regional lymph node metastases.27 Fine needle aspiration biopsy and calcitonin staining can provide accurate diagnosis. Basal serum calcitonin is high in patients with palpable MTC, and correlates with tumor burden; raised calcitonin levels following tumor removal indicate persistent or recurrent disease.7,30–33 Exceedingly high calcitonin levels indicate that the disease is disseminated and that patients cannot be cured by surgery. Family screening for hereditary MTC have previously utilized pentagastrin (now more often calcium) to stimulate calcitonin secretion from malignant or hyperplastic C cells. Normal basal but raised stimulated calcitonin values are claimed to occur with CCH, but the stimulation tests may not always safely distinguish between CCH and minimal MTC.7,10 Occasionally, nonfunctioning pancreatic endocrine tumors secrete calcitonin and cause raised serum calcitonin values, but values then fail to rise after stimulation as expected with MTC.34 Carcinoembryonic antigen (CEA) is often used as an additional tumor marker, but is not specific for MTC, and does not always follow calcitonin values.7,35
In MEN 2A, nearly 90% of patients develop MTC with penetrance dependent on the affected RET gene codon.7,10,30,36,37 Unilateral or bilateral typically multicentric PHEO develop in altogether 50% of patients, and PHPT in 15% to 30%. MEN 2A is the most common of the MEN 2 syndromes comprising 50% to 60% of patients with hereditary MTC. MTC is generally the first manifestation of MEN 2A and develops with strong genotype-phenotype correlation at a variable age dependent on the involved codon of the RET gene (Figs. 48-1 to 48-3). In 95%, MEN 2A occurs as a result of mutations in exon 10 or 11 codons, approximately 40% of patients caused by the classical codon 634 mutations. This is depicted as the ATA class C mutation in the recent American Thyroid Association (ATA) Guidelines.37 MEN 2A is also caused by exon 10 mutations in codons 620, 618, less often codons 611 and 609 (all ATA class B), and exceptionally by mutations affecting codons 768, 790, 791, 804, or 891 (ATA class A mutations)8–12,36,37 (Figs. 48-1 and 48-2). With the ATA-C (codon 634) mutations, MTC has been reported as early as 1 year of age, with ATA-B mutations at 5 years, and with ATA-A mutations at 10 years.7,27,30,33,36,37
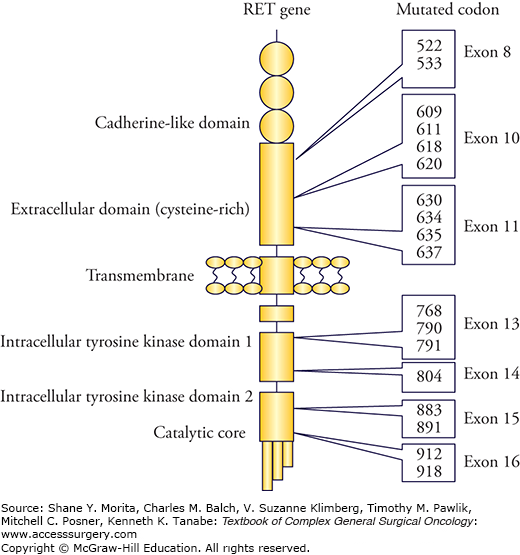
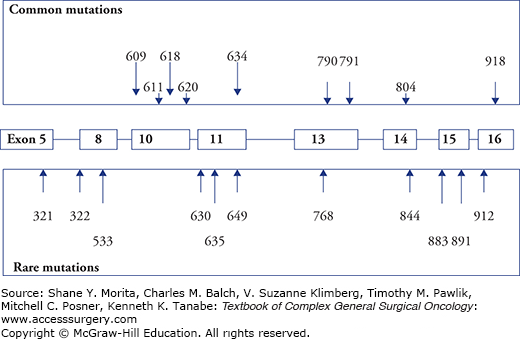
FMTC has comprised 35% to 40% of MEN 2 cases without PHEO or PHPT despite presence of the typical MEN 2 germline mutations.15 Conversion to the MEN 2B phenotype will virtually invariably occur during follow-up of patients with high penetrance mutations in codon 918, and to MEN 2A phenotype with the codon 634 mutations. The same conversion to MEN 2A phenotype will often occur with mutations in codons 620 and 618, less often for mutations in codons 611 and 609, and rarely for low penetrance mutations in codons 768, 790, 791, 804, and 891.7,10,30 FMTC has been increasingly diagnosed when genetic screening has been more generally applied, and is now mainly considered as a variant of MEN 2A rather than a distinct entity.7,10,37 Classifying as FMTC should only be done after follow-up of a large number of family members from several generations, due to risks to miss MEN 2A and rare presence of PHEO with some mutations.7,10,11,27 When follow-up has been long enough, fewer mutations have appeared to be associated with only FMTC.
Germline mutations causing paradoxal RET gene inactivation with loss of function (with congenital absence of the enteric innervation) have been implicated in 10% to 40% of cases with Hirschsprung’s disease (HD) without MEN 2A/FMTC.7,17,30,37–42 In 2% to 5% HD has also appeared with ATA class B activating RET gene mutations (codons 620 and 618, more rarely 611 and 609, and occasionally with ATA class A codon 791 mutations) causing MEN 2A/FMTC with low penetrance. In these cases, HD has often preceded clinical manifestations of MTC and PHEO.
Cutaneous lichen amyloidosis, a pruritic lichenoid skin lesion in the scapular region corresponding to dermatomes T2 to T6 on the upper back, has been reported in 9% of patients with RET codon 634 (ATA class C) mutations (and one case with codon 804 mutation).7,30,43,44 The skin lesion has often preceded clinical manifestations of MTC, and been interpreted as a sensory neuropathy involving the dorsal spinal nerves, with secondary changes due to deposition of amyloid due to repeated itching.7,9,10,12,43,44
MEN 2B is the rarest subtype, comprising around 5% to 10% of MEN 2 cases. Patients have developed MTC at the time of birth or within the first few months, 50% develop PHEO typically around puberty, whereas PHPT is not encountered.7,10,16,18–20,36,37,45–51 MEN 2B is in 95% to 98% caused by codon 918 mutations (ATA class D), and in 2% to 3% by mutations in codon 883 (also ATA class D) with apparently less aggressive MTC and occasional cases with long-term cure.7,10,16,18–20,45–51 Double (tandem) mutations involving codons V804M and either codon E805K, Y806C, or S904C have been characterized by later age of onset and varying aggressiveness of the MTC.52–55
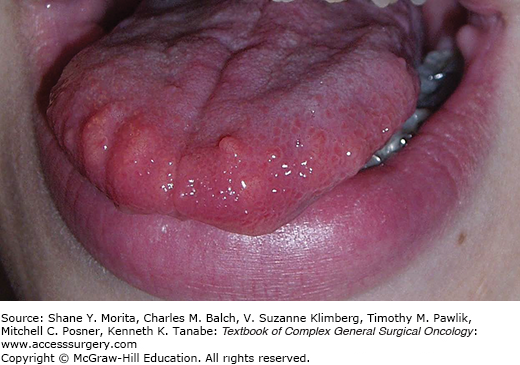
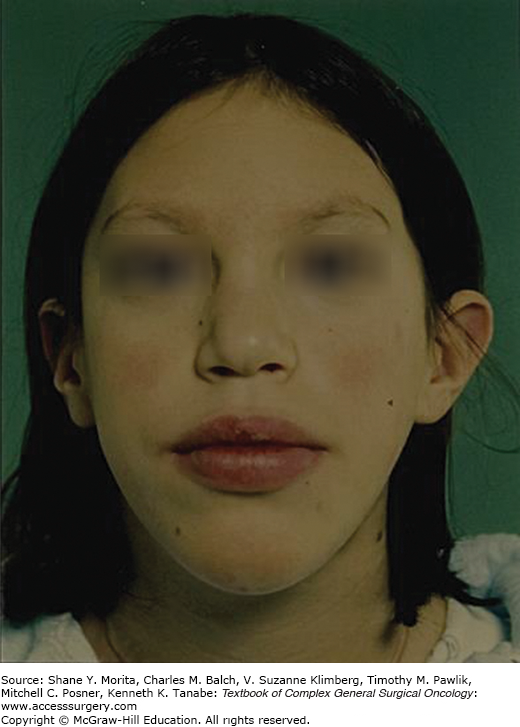
Nonendocrine manifestations of MEN 2B cause the characteristic phenotype, with typical faces, large lips, prominent phrenulum, dental irregularities, and presence of characteristic bumpy mucosal nodules due to neuromas in the tongue, lips, and buccal mucosa7,10,30,45–51,56,57 (Fig. 48-4). The patients may also have neuroma nodules in the conjunctiva, conjunctivitis sicca with inability to cry tears, thickened corneal nerve fibers, and entropium (infolding of eyelid margins).30 More than 90% have abdominal symptoms due to diffuse ganglioneuromatosis in the gastrointestinal tract with obstipation, diarrhea, or bloating, and ultimately development of megacolon in two thirds of the patients (“pseudo Hirschsprung’s disease”).56–58 Characteristic is also an asthenic Marfanoid body habitus with long bowing extremities and hyperflexible joints, pectus excavatum, scoliosis, epiphysiolysis, and proximal muscle weakness.7,10,45,48 However, neurotrophic effects may be seen as thickened nerve sheaths, and prominent recurrent laryngeal nerves at neck operations.30 Patients with MEN 2B have early MTC onset during the first months of life and aggressive thyroid cancer with early dissemination and dismal prognosis. Prophylactic thyroidectomy is rarely possible because the majority of patients appear as de novo cases without positive family history due to spontaneous new RET mutations.7,10 The patients “look different than other family members”, but have often experienced delay in diagnosis until palpable thyroid tumors, or symptoms of PHEO have become obvious. The syndrome may be recognized in infants by failure to thrive due to suckling weakness and chronic obstipation, and by the presence of characteristic oral mucosal neuromas. As importantly emphasized, the typical “crying without tears” due to conjunctivitis sicca and reduced tear production may be recognized by parents and physicians before other features become obvious.7,10,30,46–48,56–58
The recently revised ATA Medullary Thyroid Cancer Guidelines emphasize that RET mutations are associated with nearly complete penetrance of MTC phenotype at young age.7,37 When metastases have occurred the patients are often incurable with high morbidity and mortality, which can be minimized by early intervention. Proposed strategies for timing of prophylactic thyroidectomy during childhood have considered age cut-offs based on the youngest age MTC has developed or metastatic disease occurred.7,30,37 Thyroidectomy prior to dissemination with lymph node metastasis can obviate the need for central compartment lymph node dissection with the inherent increased risk of causing hypoparathyroidism (and vocal cord paralysis).
The guidelines substantiate that patients with germline RET mutations should undergo prophylactic thyroidectomy based mainly on RET gene testing, as this has lower rate of false indications than calcitonin testing (exceptions could be considered with the lowest risk mutations).7,10,30,34,36,37 The guidelines depict the highest risk with ATA class D mutations at codons 918 and 883 (MEN 2B phenotype), associated with MTC onset at time of birth and highest risk of metastases and disease-specific mortality (Table 48-1).7,10,30,34,36,37 ATA class C mutations at codon 634 have had MTC onset as early as age 1 year, and have lower, but still high risk of early and aggressive MTC. ATA class B mutations at codons 609, 611, 618, 620, and 630, have had MTC at age 5 years and carry lower risk for aggressive MTC. ATA class A mutations at codons 768, 790, 791, 804, and 891 have had MTC at age 10 years and have the lowest risk, but have been a heterogeneous group, some have MTC with late onset, incomplete penetrance, and high biochemical cure rate when subjected to prophylactic thyroidectomy at ages older than 5 years.7,30,36,37,59–62 However, ATA class A RET mutations also comprise an unpredictable minority with aggressive MTC, as in a 6-year-old proband with metastatic MTC due to an 804 RET mutation.37,60–62 Mutations at codon 804 were initially thought to be associated only with FMTC, but subsequent studies identified patients with late-onset PHEO and PHPT (associated with 804 L but not 804 M mutations).7,60,61 The same applies to codon 609 mutations where initial studies had revealed no cases of PHEO, but subsequently PHEO was detected as the dominating endocrinopathy in one family, and the onset of MTC varied from 9 years in one kindred to 48 years in another.7 Recently, exon 8 G533C mutations, which had been linked only to FMTC, were found to include cases of MTC and PHEO and could therefore also be associated with MEN 2A.7
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


