Fig. 12.1
The PI3K pathway and associated inhibitor classes. Schematic representation of the PI3K/Akt/mTOR pathway (red) and the MAPK pathway (blue) including recognized points of crosstalk between the respective cascades. PI3K is represented by the heterodimer of p110 and p85. The pathway elements mutated in human cancer are marked with a yellow star. The classes of inhibitors in clinical development are represented in purple
Given that the physiologic endpoints of PI3K activation, when unchecked, lead to the several of the hallmarks of cancer, it is not surprising that the pathway is also central to many aspects of the malignant process. Further, genetic phenomena that lead to constitutive pathway activation are common in human cancer. In addition to activating mutations or amplifications in RTKs (HER2, EGFR, ALK, MET, etc), the most relevant alterations are mutations and amplifications within the catalytic subunit of PI3K (p110-alpha, coded for by the PIK3CA gene) and loss of function of the PTEN tumor suppressor. The frequencies of these aberrations activating the pathway across different tumor types are summarized in Fig. 12.2. Mutations also affect other components of the pathway less commonly including Akt, TSC, and LKB1. Consequent to its importance and its frequent genetic deregulation in cancer, the PI3K pathway has become an attractive target for developmental therapeutics in oncology. The first compounds on the scene were the rapalogs.
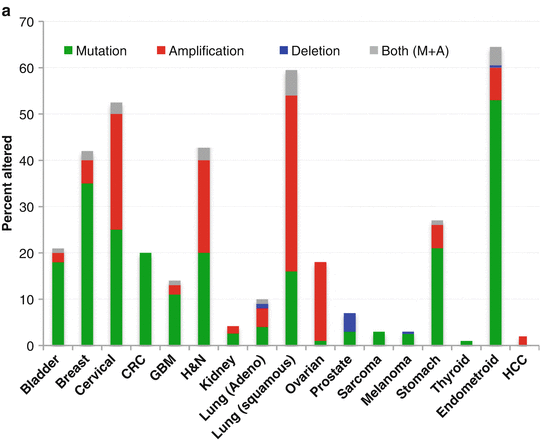
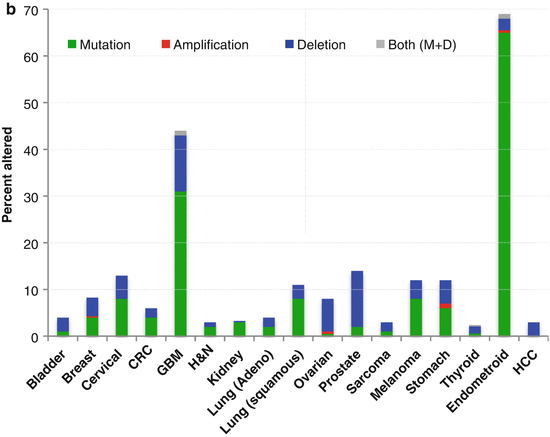
Fig. 12.2
Genetic aberrations in (a) PIK3CA and (b) PTEN genes across common tumor types. Data represented as a percentage of total cases evaluated (Data obtained from The Cancer Genome Atlas at http://www.cbioportal.org/public-portal/). CRC colorectal, GBM glioblastoma multiforme, H&N head and neck, HCC hepatocellular carcinoma
Rapamycin, the prototype agent of this class, acts by binding to the cytosolic protein FK-binding protein 12 (FKBP12); the resultant complex in turn allosterically inhibits mTOR by directly binding to the mTORC1 complex. The clinical experience with rapalogs extends back to the use of rapamycin as an immunosuppressive agent to prevent organ rejection. A serendipitous observation of regressing dermal lesions of Kaposi’s sarcoma in renal transplant patients being treated with rapamycin diverted attention to these agents being used as anticancer agents. The administration of rapalogs in oncology has met with success, albeit modest as single agent, and many combination strategies continue to undergo clinical investigation (see Chap. 11).
Building on these earlier successes, combined with a growing understanding of the PI3K pathway and its biological relevance to the malignant process, a battery of new molecules from several drug classes targeting key nodes in this critical signaling pathway are emerging. Many such agents have reached clinical evaluation in early phase trials, with a host of completed monotherapy studies and a growing list of combination studies. Here, we review the key properties of these inhibitors, pertinent findings from the trials, and consider some of the challenges facing the development of these agents.
12.2 Dual PI3K/mTOR Inhibitors
The allosteric inhibition of mTOR by the rapalogs leaves mTORC2 largely unaffected and also results in only partial inhibition of the mTORC1 substrate 4EBP1 [2, 3]. In comparison, the catalytic mTOR-targeted therapies not only affect both mTOR complexes, but the level of mTORC1 suppression is more complete [4]. The dual PI3K/mTOR inhibitors are so named due to their ability to inhibit both the mTORC complexes in addition to the class I PI3K isoforms in an ATP-competitive manner. The dual nature of their activity stems from the structural similarity of the ATP-binding pocket in the catalytic domain of both mTOR and the p110 subunit of PI3K. By targeting the PI3K pathway at two key nodes, it offers a theoretical advantage of achieving more profound pathway inhibition, broadens the spectrum of genotypes that may be sensitive to the drug, and prevents intra-pathway deleterious compensatory signaling [5]. Examples of dual inhibitors, most of which are orally administered, include BEZ235 (Novartis), XL765/SAR254409 (Exelixis/Sanofi), GDC-0980 (Genentech), PF-05212384 (Pfizer), and GSK2126458 (GlaxoSmithKline).
12.3 mTORC1/2 Inhibitors
The discovery that mTORC2 plays a direct role in the activation of Akt, combined with limitations in the clinical antitumor activity of the rapalogs and the consequences of feedback loops, has led to the development of ATP-competitive inhibitors of mTOR kinase. Similar to the dual PI3K/mTOR inhibitors, the mTORC1/2-targeted therapies are catalytic inhibitors of both mTORC1 and mTORC2 complexes. This allows for inhibition of phosphorylation of Akt on the rapamycin-insensitive mTORC2-dependent site in addition to a more profound effect on mTORC1 [6]. They differ from the dual PI3K/mTOR inhibitors by sparing PI3K from their effects. Theoretically, inhibiting fewer targets may be associated with an improved toxicity profile. MLN128 (Millennium), OSI-027 (OSI Pharmaceuticals), AZD2014 and AZD8055 (AstraZeneca), and CC-223 (Celgene) are relevant examples.
12.4 Pan-PI3K Inhibitors
The pan-PI3K inhibitors selectively target the class I PI3K isoforms while sparing mTORC1/2 from inhibition. They are predominantly orally administered agents and ATP competitive. Further, members of this group target both mutant forms of PI3K-alpha as well as the wild-type beta, delta, and gamma isoforms. There is rationale to this approach, because although PI3K alpha is the most relevant to human cancer with its frequent mutations and amplifications, accumulating evidence is implicating the other isoforms in malignant processes, even if oncogenic mutations in these elements are not described [7]. Preclinical evidence suggests that the pan-PI3K inhibitors show greatest sensitivity in a context of upstream RTK or PI3K activation due to of ERBB2 amplification or PIK3CA mutation, respectively [8, 9]. In contrast, the presence of intrinsic pathway activity driven by factor downstream of mTOR or in parallel pathways (such as KRAS mutation) is less likely to derive benefit from these agents [10, 11]. Examples of pan-PI3K inhibitors include BKM120 (Novartis), XL147/SAR245408 (Exelixis/Sanofi), GDC-0941 (Genentech), CH5132799 (Chugai Pharmaceutical), and BAY 80–6946 (Bayer).
12.5 Isoform-Specific PI3K Inhibitors
One of the concerns of targeting all class I PI3K isoforms, with or without concomitant mTOR inhibition, is that the high number of drug targets has the potential to increase toxicity. Accordingly, isoform-specific PI3K inhibitors are in development, with the intent of maximizing therapeutic benefit while minimizing undesirable side effects. These inhibitors are being explored in more restricted genetic contexts. Examples include the p110-alpha inhibitors BYL719 (Novartis) and MLN1117 (Millennium), the so-called p110-beta-sparing GDC-0032 (Genentech), the p110 beta inhibitor GSK2636771 (GlaxoSmithKline), and the p110 delta inhibitors idelalisib (formerly GS 1101 and CAL-101 (Gilead/Calistoga)) and AMG319 (Amgen). Similar to the pan-PI3K inhibitors, greatest sensitivity to the PI3K-alpha inhibitor BYL719 was found in cells with PIK3CA mutation or ERBB2 amplification; conversely, PTEN and BRAF mutations were associated with resistance to this drug [12]. Other preclinical research identified the vulnerability of PTEN-deficient tumors to PI3K-beta inhibition [13, 14]. Finally, PI3K-delta expression is restricted largely to hematopoietic cells where it plays a critical role in B-cell homeostasis and function via its capacity to integrate signal downstream of surface receptors including the B-cell receptor [15]. PI3K delta also promotes malignant B-cell proliferation and survival, which is abrogated by the administration of PI3K-delta-specific inhibitors, prompting clinical development [16, 17].
12.6 Akt Inhibitors
Akt is a central hub in the PI3K pathway that has drawn the interest of researchers as an alternate druggable target. There are three Akt isoforms (Akt1/2/3) that can be inhibited collectively or specifically. The former is achieved by means of catalytic inhibition that targets the ATP-binding pocket. However, the ATP-binding pocket shares sequence homology with other kinases such as p70S6K leading to specificity concerns. Conversely, Akt1/2 isoform-specific inhibition occurs via an allosteric mechanism whereby binding to the pleckstrin homology domain prevents Akt membrane localization [18, 19]. The potency of the allosteric inhibitors is negatively affected by the presence of Akt-activating mutations, such as the E17K mutation in Akt1 found in human cancers [20]. Conversely, ERRB2 amplification, PIK3CA mutations, or PTEN loss of expression predicts for heightened sensitivity to these agents in preclinical models [21–23]. Members of this drug class include MK-2206 (Merck), GDC-0068 (Genentech), GSK2141795 (GlaxoSmithKline), and AZD5363 (AstraZeneca).
12.7 Safety and Toxicity
The safety profile of non-rapalog agents targeting the PI3K signaling cascade has been generally acceptable, with toxicities largely reported as mild to moderate in severity, reversible, and manageable. The common drug-related adverse effects appear to be quite consistent across the drug classes, and many of these also account for the dose-limiting toxicities (DLTs). Broadly speaking, constitutional symptoms (fatigue and asthenia), cutaneous toxicities (primarily rash), gastrointestinal complaints (anorexia, nausea, vomiting, dyspepsia, and diarrhea), stomatitis (or mucositis), and hyperglycemia have been prevalent. The lipid profile alterations frequently seen with rapalogs have not been encountered.
Constitutional symptoms, variably reported as lethargy, fatigue, and asthenia, have featured in the adverse effect profile across all described drug classes, as have the gastrointestinal complaints, with nausea and diarrhea being particularly prominent. Due to the typically chronic administration of these compounds, even mild to moderate side effects (the majority of cases for these toxicities) can have a significant negative impact on the quality of life of patients and influence drug tolerability leading to dose reductions, interruptions, or cessations. Careful monitoring and early treatment of toxicities are essential in order to reap the benefits of their antitumor efficacy.
Mucositis, perhaps not as prevalent in reported trials when compared with rapalogs, has been described with a number of the inhibitors targeting this signaling cascade. Its importance is underscored by the fact that it was dose limiting in trials of the dual PI3K/mTOR inhibitors PF-05212384 and BEZ235 (the latter when dosed twice daily) [24, 25], the mTORC1/2 inhibitors MLN0128 and CC-223 [26, 27], as well as the Akt inhibitor MK-2206 [28].
Rash was dose limiting for several of the dual PI3K/mTOR inhibitors (GDC-0980, PF-05212384) [25, 29] and the pan-PI3K inhibitors (BKM120, XL147, GDC-0981) [30–33]. Rash has also been quite problematic among the Akt inhibitors, such as MK-2206 and AZD5363 [28, 34]. The rash observed with these agents has been described as erythematous, non-blistering, and maculopapular. This is distinct from the acneiform rash observed with EGFR-targeted agents, but shares characteristics with the papulopustular or maculopapular rapalog-induced rash that occurs in almost 30 % of patients [35].
Thus far, the development of pneumonitis has not been as problematic with newer agents when compared with the rapalogs. However, it was observed as a DLT with the dual PI3K/mTOR inhibitor GDC-0980 in its first-in-human dose-escalation study, and steroid-responsive interstitial pneumonitis was also described in two patients on the phase I study of the pan-PI3K inhibitor BAY 80-6946 [29, 36]. More recently, four cases were observed in patients treated with the mTORC1/2 inhibitor CC-223 [37]. Despite the relative infrequency of this important toxicity, clinicians and researches need to remain vigilant as it is a potentially life-threatening complication, and its true incidence is yet to be determined given the small numbers of patients who have been treated with these compounds.
Glucose homeostasis is influenced by the PI3K pathway. Therefore, the anticipated toxicity of hyperglycemia commands particular interest as it represents an on-target effect of these agents and a potential pharmacodynamic biomarker of pathway inhibition. As predicted, elevated blood glucose levels have been reported for many compounds, in particular at higher doses when the degree of pathway inhibition is greater. The impact on glucose metabolism has been both common (all grades hyperglycemia described in 83 % of patients treated with the dual PI3K/mTOR inhibitor GDC-0980 [29]) and dose limiting (PF-05212384, BKM120, GDC-0941, AZD5363, and BYL719) among others [25, 30, 32, 34]. Many trials have employed strict algorithms for the management of hyperglycemia. In most cases, administration of metformin has allowed for effective control of blood sugar levels, though in some instances the severity of the glucose elevation has necessitated dose reductions or subcutaneous insulin [38].
The other important metabolic consequence observed has been abnormalities in liver function, in particular elevations in alanine transaminase and aspartate transaminase. Such transaminase elevations have been described with several agents including XL765 and CAL-101 [39, 40] and was dose limiting with AZD8055 and CH5132799 [41, 42]. These elevations were typically mild to moderate, but even when more severe, they tended to be reversible and without long-term sequelae.
One somewhat unusual toxicity is the mood alteration described with the pan-PI3K inhibitor BKM120 [30]. Found to be both common and in more severe instances dose limiting, such neuropsychiatric effects of the drug imply penetration into the central nervous system which may in turn suggest potential utility of this drug for primary brain cancers or brain metastases [30].
At times, the administration schedule of a drug affects its toxicity profile. This was the case in the phase I study of AZD5363, where continuous dosing led to dose-limiting toxicities of rash and diarrhea, as opposed to an intermittent schedule where the more manageable and more tolerable adverse event of hyperglycemia was the DLT [34].
12.8 Pharmacodynamic Biomarkers
Pharmacodynamic (PD) biomarkers are markers of drug effect that assess for target inhibition and pathway downregulation. They necessitate assessment prior to and following an intervention to detect a change from baseline; a correlation with clinical activity is not implied but is desirable. The PD biomarkers applied across trials of the PI3K inhibitors have most typically been the activation status of relevant pathway nodes. Specifically, the level of phosphorylation of the following residues pre- and posttreatment gives a measure of PD activity at different levels of the pathway: Akt at residue Thr308 for PI3K activity, Akt at residue Ser473 as an mTORC2 readout, PRAS40 at residue Thr246 for Akt activity, eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1) at Ser65 and Thr70 for mTORC1 activity, and ribosomal protein S6 (RPS6) at Ser240 and Ser244 as a marker for mTORC1/S6K activity. Ki67 and TUNEL readouts have also been investigated as PD biomarkers of proliferation and apoptosis, respectively.
Consequent to the role that the PI3K pathway plays in glucose metabolism, 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) scans have been applied as a PD biomarker in many studies to date. There has been a consistent trend toward a reduction in PET avidity of tumors following drug administration across trials employing this imaging modality. However, despite these encouraging findings, it is yet to be determined whether these changes represent genuine antitumor activity or whether it is merely a bystander effect on glucose homeostasis without yielding biological relevance as an anticancer agent. With no strong or consistent correlation observed between PET-defined metabolic responses and CT-defined RECIST responses in reported trials, such results needed to be interpreted with caution.
Other metabolic PD biomarkers explored have similarly attempted to exploit the effects of the pathway in glucose homeostasis by measuring fasting levels of glucose, insulin, and C-peptide in plasma. In some instances, the information gleaned from these analyses have contributed to determination of a biologically relevant dose, but the influence of confounding factors such as diet and diurnal variations has limited their utility for decision making in individual cases [30].
The types of PD markers explored in early phase trials, together with relevant findings, are summarized in Table 12.1 and [43].
Table 12.1
Pharmacodynamic biomarkers
Type of PD BM | Agent | Finding | Refs. |
|---|---|---|---|
Skin | BEZ235 | ↓ levels of pS6 | |
XL765 | ↓ levels of pAKT Thr308, pAKT Ser473, pPRAS40, p4EBP1, and pS6 (40–90 %) | [39] | |
MLN128 | ↓ levels of p4EBP1, pS6, and pPRAS40 (60–100 %) in most pts | [27] | |
BKM120 | ↓ levels of pS6 (40–85 %) in 15 of 19 pts treated at 80–150 mg (MTD = 100 mg) | [30] | |
XL147 | ↓ levels of pAKT Thr308, pAKT Ser473, pPRAS40, p4EBP1, and pS6 (50–80 %) | [94] | |
Hair follicles | XL765 | ↓ levels of pAKT Thr308, pAKT Ser473, pPRAS40, p4EBP1, and pS6 (50–90 %) | [39] |
XL147 | ↓ levels of pAKT Thr308, pAKT Ser473, pPRAS40, p4EBP1, and pS6 (20–50 %) | [94] | |
MK-2206 | ↓ levels of pPRAS40 in most pts at MTD (60 mg) | [89] | |
Platelet-rich plasma | GDC-0980 | ↓ levels of pAKT Ser473 (>90 %) for pts treated at doses ≥16mg (MTD = 50 mg) | [29] |
AZD2014 | ↓ levels of pAKT Ser473 (60–80 %) | [87] | |
GDC-0941 | ↓ levels of pAKT Ser473 | [33] | |
CH5132799 | ↓ levels of pAKT (up to 80 %) | [42] | |
MK-2206 | ↓ levels of pAKT Ser473, pPRAS40, and pGSK3β in most pts at MTD (60 mg) | [89] | |
GDC-0068 | ↓ levels of pGSK3β (≥75 %) at doses ≥200 mg (MTD = 600 mg) in dose- and time-dependent manner | [90] | |
PX-866 | ↓ levels of pAKT (>80 %) in 4 of 10 pts at the MTD (8 mg bid) | [88] | |
Peripheral blood mononuclear cells | MNL128 | ↓ levels of p4EBP1 in most pts | [27] |
AZD2014 | ↓ levels of p4EBP1 (75 %) | [87] | |
OSI-027 | ↓ levels of p4EBP1 >60 % in most pts treated at doses ≥20 mg (doses up to 40 mg presented, no MTD) | [86] | |
C-peptide | BEZ235 | Dose-dependent ↑ in plasma C-peptide | |
BKM120 | Dose-dependent ↑ in plasma C-peptide (with associated ↑ in BGL at higher doses) | [30] | |
BYL719 | Dose-dependent ↑ in plasma C-peptide | [95] | |
FDG-PET | BEZ235 | Metabolic PR in 8 of 37 pts with qd dosing and 4 of 9 pts with bid dosing | |
GDC-0980 | Metabolic PR in 5 of 6 pts | [29] | |
BKM120 | Metabolic PR in 9 of 19 pts | [30] | |
GDC-0941 | Metabolic PR in 6 of 17 pts | [33] | |
CH5132799 | Metabolic PR in selected cases | [42] | |
BYL719 | Metabolic PR in 10 of 17 pts | [95] | |
GDC-0032 | Metabolic PR in 7 of 13 pts | [48] | |
Tumor tissue | BEZ235 | ↓ levels of pS6 and ↓ Ki67 (selected cases) | |
XL765 | ↓ levels of pAKT Thr308 (50–75 %), p4EBP1 (60–80 %), and pERK (50–80 %) in 5 pts at the MTD (50 mg bid) | [83] | |
AZD2014 | ↓ levels of pS6, pAKT Ser473, and Ki67 (selected cases) | [87] | |
XL147 | ↓ levels of pAKT Thr308 (40–80 %), p4EBP1 (50–70 %), and pERK (40–60 %) in 9 pts at the MTD (600 mg) | [31] | |
MK-2206 | ↓ levels of pAKT Ser473 in all 12 pts at MTD (60 mg) with 9 of 12 >50 % and 4 of 12 >90 % reduction | [89] | |
GDC-0068 | ↓ levels of pPRAS40 (60–70 %) and cyclin D1 (50 %) in 3/3 pts treated at 400 mg (MTD = 600 mg) | [90] |
12.9 Efficacy
As previously described, the PI3K pathway is an attractive target for anticancer therapies due to its role in malignant processes and its widespread activation in human cancer. Further, much work has gone toward overcoming early drug selectivity issues as evidenced by the large number of compounds now in development. It has therefore been with much optimism and expectation that these agents entered early phase clinical trials.
The most impressive results to date have occurred in the hematological malignancies. Administration of PI3K-delta isoform-specific inhibitors has been explored in the clinic because these cancers depend on PI3K-delta signaling. Use of single-agent idelalisib in separate phase I trials of chronic lymphocytic leukemia, indolent non-Hodgkin lymphoma, and mantle cell lymphoma, all in a relapsed or refractory setting, yielded spectacular response rates of 56 %, 48 %, and 40 %, respectively [44–46]. Later phase clinical trials conducted on the basis of these results have led to the approval of idelalisib in both the USA and Europe for relapsed CLL (in combination with rituxumab) and relapsed follicular lymphoma, and in the USA for relapsed small lymphocytic lymphoma.
Bearing in mind that efficacy is not a primary objective of phase I studies, the single-agent PI3K pathway inhibitor clinical trials in solid tumor types have been somewhat disappointing overall. The most encouraging results have occurred with use of PI3K-alpha isoform-specific inhibitors; separate phase I studies of BYL719 and GDC-0032 have seen partial responses in 9 % and 15 % of patients, respectively [47, 48]. However, many patients have now been treated on monotherapy studies employing agents of varying class and mechanism. Despite the numbers treated (more than 1500 patients in total), relatively few responses have been documented thus far. Where results in solid tumors are available, taking into account that some results are preliminary or the trials are incomplete, it appears that only about 2–3 % of patients have shown radiological RECIST reported responses to date (Table 12.2).
Table 12.2
Reported responses from monotherapy studies of inhibitors of the PI3K pathway including tumor types and relevant genetic aberrations where known
Drug | Pts –total | Responses | PI3K pathway activation | Other pathway activation | No known pathway activation | Unknown/not reported | Reference |
|---|---|---|---|---|---|---|---|
Dual PI3K/mTOR inhibitors | |||||||
SF1126 | 44 | 0 | [81] | ||||
BEZ235 | 100 | 2 | NSCLC (PTEN mut) | ER+ breast | |||
XL765 | 79 | 0 | [83] | ||||
GDC-0980 | 42 | 1 | Adrenocorticala | [29] | |||
PF-04691502 | 30 | 0 | [84] | ||||
PF-05212384 | 47 | 0 | [25] | ||||
GSK2126458 | 129 | 4 | RCC (PTEN loss) Bladder (PIK3CAmut) | RCC | Bladder | [85] | |
mTORC1/2 inhibitors | |||||||
MLN128 | 52 | 1 | RCC | [27] | |||
OSI-027 | 43 | 0 | [86] | ||||
AZD8055 | 49 | 0 | [41] | ||||
AZD2014 | 54 | 1 | Pancreas | [87] | |||
CC-223 | 129 | 5 | ER+ breast NSCLC 3× HCC | ||||
Pan-PI3K inhibitors | |||||||
BKM120 | 35 | 1 | TNBC (KRAS mut) | [30] | |||
XL147 | 78
Related posts: The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
The Role of PI3K/AKT/mTOR Inhibitors in the Treatment of Hematological Malignancies
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
The Role of PI3K/AKT/mTOR Inhibitors in the Treatment of Hematological Malignancies
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors
 Predictive Biomarkers of Response to mTOR Inhibitors Predictive Biomarkers of Response to mTOR Inhibitors
 Rational Combinations of mTOR Inhibitors as Anticancer Strategies Rational Combinations of mTOR Inhibitors as Anticancer Strategies
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||||||