Ag category
Examples
Expression in cancer
Tissue differentiation Ags
Mclan-A/MART-1, gp100, tyrosinase, TRP-1, TRP-2
Melanoma
PSA
Prostate carcinoma
Prostate-specific membrane Ag (PSMA)
Prostate carcinoma
MUC-1
Particular adenocarcinomas
MUC-16 (CA-125)
Mainly ovarian cancer and also in endometrial cancer, fallopian tube cancer, lung cancer, breast cancer, and gastrointestinal cancer
EpCAM
Various carcinoma types
Gangliosides (GM2, GD2, GD3)
Melanomas, small cell lung cancer, and neuroblastoma
CD5
T-cell leukemia/lymphoma
CD19, CD20, CD21, CD25, CD37
B-cell lymphoma
CD30
Hodgkin lymphoma
CD33, CD45
Acute myeloblastic leukemia
CAMPATH-1 (CDw52)
Lymphoid malignancies (T and B cell)
Oncofetal Ags
CEA
Expressed on several gastrointestinal malignancies and adenocarcinomas
AFP
Hepatocellular carcinoma, germ cell tumors, and metastatic cancers of the liver
β-hCG
Germ cell tumors and choriocarcinoma
Cancer-testis Ags
MAGE 1, 3, 12, NY-ESO, BAGE, GAGE, LAGE
Various tumors
Viral Ags
Human papillomavirus 16 E6 and E7 proteins
Cervical and anal cancers
Growth factor receptors
EGFR
Lung, glioma, breast, head, and neck tumors
ERBB2
Breast, ovarian, stomach, and endometrial carcinoma
CD140b (PDGFRB)
Various tumor types
Stromal Ags
Fibroblast activation protein (FAP)
Colon, breast, lung, head, and neck carcinoma
Tenascin, metalloproteinases
Colon, breast, lung, head, and neck carcinoma
Vascular Ags
Endosialin
Breast cancer, colon carcinoma, neuroblastoma
Vascular endothelial growth factor (VEGF)
Metastatic colorectal cancer, NSCLC, metastatic breast cancer, glioblastoma, metastatic renal cell carcinoma
αVβ3
Melanoma and prostate cancer
16.4.3 Target Identification Approaches
Several efficient methods have been promoted to identify the potential differences between tumor and non-tumor cell lines and/or tissues at the DNA, mRNA, protein or Ab reactivity levels. Several major techniques used for the discovery of tumor antigens are briefly described below.
16.4.3.1 Genomics
Cancer-related alterations in genome include silent mutations (e.g. deletions and insertions) [46, 47], gene amplification [48] and larger scale defects such as chromosomal translocations [49]. Today, gene amplifications or deletions as well as chromosomal translocations are detected using several techniques such as comparative genomic hybridization (CGH) [50, 51] and spectral karyotyping (SKY) [52–54]. Amplification of HER2 gene is known as the first solid tumor-associated genomic aberration, which led to the successful development of trastuzumab [55].
16.4.3.2 Transcriptomics
Two approaches commonly employed to analyze global gene expression in tumors include microarray analysis and serial analysis of gene expression (SAGE). Microarray is based on the hybridization of fluorescently-labeled sequences (probes or targets) to their complementary sequences [56, 57]. Complementary DNA (cDNA) microarray has been used to identify the frequency of elevated tumor Ag expression, for instance, in acute myeloid leukemia (AML) [58]. In 1995, Velculescu et al. [59] described SAGE as a sequencing-based method for gene expression profiling, which facilitated the global and quantitative characterization of a transcriptome.
Although DNA microarray is an excellent method for rapid screening of large numbers of samples and genes, it can only examine the already-indentified sequences. In contrast, SAGE does not require prior knowledge, and represents an unbiased, comprehensive representation of transcripts [60]. Furthermore, SAGE can quantitatively identify low-abundance transcripts and detect relatively small differences in their expression [61]. Nonetheless, it is expensive and time-consuming [62] and requires relatively high amounts of RNA samples [63].
16.4.3.3 Proteomics
Genomic and transcriptomic analyses are indirect methods of protein identification and the number of transcripts identified by these methods does not necessarily correlate with protein levels [64–67]. In contrast, proteomics can be used as a direct method of searching for cancer-specific Ags. An additional advantage of proteomics is that it can identify differences in post-translational modification (PTM), a potentially important source of tumor Ags formation.
Proteomic evaluations were initiated by two dimensional gel electrophoresis and subsequent mass spectroscopy (2DE/MS) [68] and were expanded to more advanced methods. 2DE/MS has been widely used for separation of proteins in complex mixtures according to their molecular weight and isoelectric points; and identification of proteins that are differentially expressed in various malignances [69–74]. However, a major drawback of this technique is its inability to provide high throughput.
Other techniques that are used for the expression analysis of proteins include matrix-assisted laser desorption-ionization time-of-flight mass spectrometry (MALDI-TOF-MS) (used for investigation of haptoglobin expression in ovarian cancer) [75]; surface-enhanced laser desorption/ionization-time-of-flight/mass spectrometry (SELDI-TOF-MS) (used to study the association of cytosolic ubiquitin and ferritin light chain levels in breast cancer prognosis) [76]; liquid chromatography combined with tandem MS (LC–MS–MS) (used for phosphoproteomic analysis of HeLa cells at various stages in the cell cycle) [77]; and more-quantitative techniques such as isotope-coded affinity tags (ICATe) (used to identify differences in specific protein expression between nipple aspirate fluid samples from tumor-bearing and disease-free breasts) [78]; and isotope tags for relative and absolute quantification (iTRAQe) (utilized for identification of serum biomarkers in metastatic prostate cancer) [79]. Despite the advantages of these methods in identification of low molecular weight and low-abundance protein fractions of the proteome, they fall short of identifying protein-protein interactions.
16.4.3.4 Antibody-Based Technologies
Protein microarray is a high-throughput gel-free method with a tremendous potential to explore the interactions, activities and functions of proteins. This approach is divided into two major classes: (i) forward-phase arrays (FPAs) in which Abs are arrayed and probed with cell lysates, and (ii) reverse-phase arrays (RPAs), where cell lysates are arrayed and probed with Abs [80, 81]. Protein microarray has been utilized to recognize cancer-associated glycan variations on the proteins musin-1 (MUC1) and carcinoembryonic antigen (CEA) in the sera of pancreatic cancer patients [82] or to indentify biomarkers of bladder cancer [83].
Serological expression cloning (SEREX) was developed to combine serological analysis with Ag cloning techniques to identify human tumor Ags that elicit high-titer IgG [84]. SEREX is now being used for screening the sera of patients to detect a large range of different solid [85–88] and hematological malignancies [89, 90]. Moreover, SEREX in combination with two dimensional polyacrylamide gel electrophoresis (2D-PAGE) technology created a serological proteome analysis (SERPA) technique [91] through which investigators were able to identify melanoma [92], breast [93] and colorectal cancer Ags [94].
16.5 Molecular Mechanisms Involved in Monoclonal Antibody-Based Therapy
In general, Ab-based approaches are able to damage tumor cells through three mechanisms: direct elimination of tumor cells, indirect immune-mediated targeting of cancer cells, and the targeting of tumor stroma and vasculature system [2].
16.5.1 Direct Tumor Cell Elimination
Growth factor receptors that are overly expressed on tumor cells have been targeted by many therapeutic Abs that act through the blockade of ligand binding and/or abrogation of signal transduction [95]. Epithelial growth factor receptor (EGFR) family members have been the focus of several studies. For instance, HER2 is a member of the EGFR family with no identified ligand and Abs targeting this molecule have been shown to prevent receptor dimerization [96]. Trastuzumab, that is applied to the treatment of invasive breast cancers with overexpression of HER2, acts through prevention of receptor dimerization, along with activation of immune responses [97]. Moreover, Pertuzumab, another anti-HER2 mAb, has been shown to bind to a site different from that of trastuzumab and inhibit receptor dimerization [98]. Notably, a combination of trastuzumab and pertuzumab has shown promising antitumor results in preclinical models [99]. Cetuximab, a chimeric EGFR-specific mAb, could inhibit ligand binding and prevent receptor dimerization [100]. Further efforts are underway to target similar molecules such as HER3 and HER4 [101, 102].
The receptor tyrosine-kinase-like orphan receptor 1 (ROR1) has been suggested as a survival factor for certain cancers such as chronic lymphocytic leukemia (CLL) [42, 103], lung cancer, adenocarcinoma [104] and breast cancer [105]. Ab targeting of this transmembrane receptor by several studies has culminated in tumor cell elimination through the induction of apoptosis and necrosis [106–108]. A very recent study showed the role of ROR1 in survival of melanoma cell lines. Utilization of anti-ROR1 mAbs in this research could effectively induce apoptosis in the cell lines, proposing ROR1 as a potential target for future melanoma therapies [109].
16.5.2 Harnessing the Potential Capacity of Immune System to Eliminate Tumors
Due to their indispensible antitumor roles, immune responses have long been the focus of many Ab-based therapeutic strategies. The so far designed mAbs exert their antitumor effects through various immune-mediated mechanisms: ADCC, CDC, promoting Ag cross-presentation and targeting of immunomodulatory receptors (Fig. 16.1).
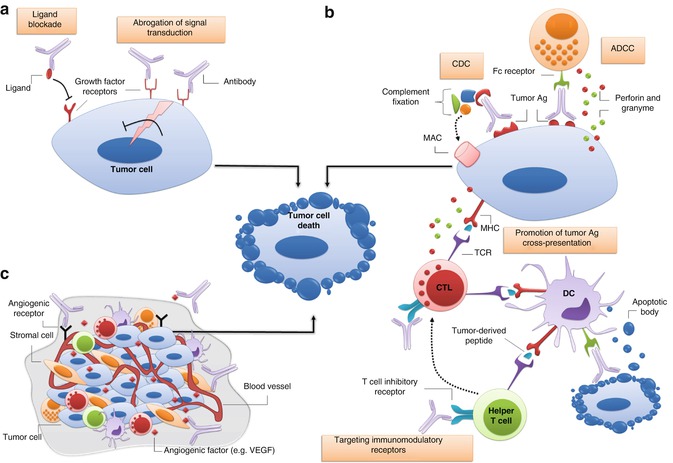
Fig. 16.1
Major mechanisms of tumor cell elimination by monoclonal antibodies. (a) Direct elimination of tumor cells is often elicited by abrogation of signal transduction via growth factor receptors (e.g. members of the epithelial growth factor receptor family) and/or blockade of ligand-receptor binding. (b) Indirect killing of tumor cells can be achieved through binding of activatory Fc receptors on immune effector cells (e.g. natural killer cells) to the Fc portion of antitumor antibody promoting antibody-dependent cell-mediated cytotoxicity (ADCC); or activation of complement compartments on the F c fragment of antibody leading to formation of membrane attack complex (MAC) and tumor cell osmotic lysis. Additionally, antibody-coated apoptotic tumor cells or apoptotic bodies that are produced following ADCC can be engulfed and presented by dendritic cells (DCs) to tumor-specific T cells. Antibodies blocking T cell inhibitory receptors (e.g. CTLA-4 and PD-1) or those stimulating activatory T cell receptors (not shown) can also indirectly improve the outcome of antitumor responses. (c) Monoclonal antibodies can also be used to antagonize receptors or ligands of tumor vasculature system, and/or to target tumor stromal cells and their products. Ag antigen, CDC complement-dependent cytotoxicity, CTL cytotoxic T lymphocyte, MHC major histocompability complex, NK natural killer
16.5.2.1 Antibody-Dependent Cell-Mediated Cytotoxicity
FcγR-dependent interactions are known to induce either stimulatory or inhibitory signals. FcγRIIIa as an activating receptor is expressed by dendritic cells (DCs), macrophages, natural killer (NK) cells and neutrophils, and is essential for NK-mediated ADCC [110]. There is an ensemble of results from both murine experiments and clinical trials establishing ADCC involvement in antitumor effects of certain mAbs. The relationship between Ab treatment and ADCC was confirmed by the study showing that rituximab (anti-CD20) and trastuzumab were less efficient in FcγR-deficient mice compared to the wild-type ones [111]. Further support was provided by the study reporting high response rates to rituximab in follicular non-Hodgkin lymphoma (NHL) patients with certain polymorphisms in the FcγRIII encoding gene [112].
Notably, a recent promising approach has been to enhance ADCC through making modifications to the Fc domain of an Ab molecule. Accordingly, an anti-CD20 Ab with enhanced affinity for FcγRIIIA could significantly increase ADCC in comparison with the original Ab and rituximab [113].
16.5.2.2 Complement-Dependent Cytotoxicity
The potential capacity of IgG subclasses to activate the classical complement pathway ending in target cell lysis and immune cell recruitment has been harnessed by several studies with the aim of eliminating tumor cells. Indeed, there is compelling evidence highlighting the relationship between complement activation and therapeutic efficacy of antitumor mAbs. A preclinical therapy model showed that the antitumor impact of anti-CD20 mAb (rituximab) was thoroughly abrogated in C1q-deficient mice [114]. Consistently, complement depletion culminated in decreased protective effect of rituximab in a murine model of human B cell lymphoma [115]. The majority of so far clinically-approved antitumor mAbs have been shown to activate ADCC and the complement pathway.
16.5.2.3 Promotion of Tumor Antigen Cross-Presentation
It is well established that Ag cross-presentation by DCs plays a pivotal part in generation of T cell responses following Ab therapy. In fact, DCs can present tumor Ag-derived peptides in the context of MHC-I molecules and stimulate tumor-specific CD8+ T cells [116, 117]. The association between Ab therapy and induction of T cell immunity was demonstrated by two studies indicating that the use of mAb increased cross-presentation of tumor Ags and cytotoxic T lymphocyte (CTL) generation [118], and that cross-presentation was enhanced following the blockade of FcγRIIB, an inhibitory receptor [119].
In general, antitumor mAbs are known to promote T cell responses through two distinct mechanisms. Firstly, Ab-mediated ADCC leads to apoptotic tumor cell generation and peptides derived from these cells might subsequently be engulfed and presented to specific T cells by DCs [120]. Secondly, Ab-coated apoptotic tumor cells can be phagocytosed, through FcγRs, and sent to the cross-presentation pathway ending in effective tumor-specific T cell responses [118, 120]. However, one should bear in mind that DCs can mediate both immunostimulatory and immunomodulatory responses depending on the tumor microenvironment [121]. Thus, it is recommended to employ Ab-based antitumor strategies in combination with approaches that target suppressive agents of tumor microenvironment.
16.5.2.4 Targeting Immunomodulatory Receptors
The interaction of T cell stimulatory or inhibitory receptors with their ligands on antigen presenting cells (APCs) or certain tumor cells determines the outcome of tumor-specific immune responses [2]. Therefore, Ab-mediated targeting of receptors on T cells or the ligands of these receptors has received widespread attention by several therapeutic studies.
There is a great deal of evidence confirming the antitumor potency of mAbs blocking T cell inhibitory receptors [122]. Among these receptors, cytotoxic T lymphocyte antigen-4 (CTLA-4) has gained increasing credibility owing to the promising preclinical and clinical results. This T cell receptor suppresses activated T cells through binding to CD80 (B7.1) and CD86 (B7.2). One study showed that blocking of CTLA-4 on both effector and regulatory T cell compartments contributed to the antitumor activity of anti-CTLA-4 Abs [123].
Data obtained from preclinical studies has provided the foundation for production of two clinically-approved anti-CTLA-4 mAbs (ipilimumab and tremelimumab). Ipilimumab owes its clinical approval to a pivotal study indicating that treatment with this mAb results in improved overall survival of patients with metastatic melanoma [124]. However, one should be cautious about employing CTLA-4 blockade in general, since it has been shown to exert a series of toxic side effects called immune-related adverse effects (irAEs) [124, 125]. Likewise, blockade of another T cell inhibitory receptor (programmed death-1; PD-1) using an anti-programmed death-1 mAb (MDX-1106) has led to favorable antitumor responses [126] and additional PD-1 targeting Abs are being investigated [127, 128].
Antibodies with agonistic effects on immunostimulatory receptors have also been tested in immunotherapeutic settings. A fully human Ab specific to CD137, a T cell activating receptor, has shown encouraging antitumor efficacy in phase I clinical trials [120]. On a cautionary note, high doses of this Ab can result in toxic effects, and studies with lower less toxic doses are currently underway [2]. Encouraging results upon employing Abs with agonistic impacts on CD40 have also been noted in the literature [127].
16.5.3 Targeting Tumor Stroma and Vasculature
Factors that support angiogenesis as well as those that form the extracellular matrix play an indispensible role in tumor survival [129–131]. Therefore, targeting tumor microenvironment has been shown to be of great therapeutic value in preclinical and clinical settings [132].
Vascular endothelial growth factor (VEGF), secreted by many solid tumors, supports tumor angiogenesis by binding to its receptor on endothelial cells. A combination of chemotherapy and anti-VEGF mAb (bevacizumab) is clinically approved for therapy of patients with colorectal, breast and non-small cell lung cancers (NSCLCs) [131]. Ab-targeting of VEGF receptor (VEGFR) has also been investigated by several studies. Ramucirumab, an anti-VEGFR2 mAb, showed potential antitumor impacts in a murine cancer model [133]. Consistently, targeting of VEGFR-1 by a fully human mAb showed favorable preclinical results [134].
As for many therapeutic mAbs, the growing use of bevacizumab resulted in the emergence of bevacizumab-resistant tumors due to the upregulation of alternative angiogenic factors such as platelet-derived growth factor (PDGF), which supports the growth of blood vessels through binding to its receptor (PDGFR) [135]. In fact, the addition of an anti-PDGFR mAb to anti-VEGFR-2 therapy showed promising antitumor results in preclinical models, introducing an efficient solution for treatment of bevacizumab-resistant tumors [136].
Cancer cells often press tissue stromal cells into service to provide a more hospitable microenvironment. In addition, cancer-associated fibroblasts (CAFs), as the most frequent cell population in tumor microenvironment, have a crucial role in growth and metastasis of solid tumors. Hence, approaches that target CAFs and/or molecules secreted by them have recently gained momentum [137]. For instance, a mAb directed against fibroblast activation protein (FAP), produced by CAFs, elicited robust antitumor responses in a phase I clinical trial in patients with advanced or metastatic FAP-positive colorectal cancer and NSCLCs [130].
16.6 Engineered Antibodies
Two features of mAbs that have made them interesting drug candidates are high target specificity and organization into distinct structural and functional domains. These features have facilitated protein engineering of intact Abs by a variety of methods to suit for diverse therapeutic applications. Antibody engineering techniques have attempted to optimize the therapeutic efficacy of untouched Abs, and to overcome their shortcomings by creating novel Ab structures with features such as decreased immunogenicity, optimized stability, higher binding affinity, effective tissue penetration, modified Fc function, rapid renal clearance and ease of production. Notably, advances in molecular biology has made it possible to go beyond optimization and in fact has created entirely new Ig domain-based structures, not found in nature, which can be tailored to achieve favorable results. This section describes Ab engineering (Fig. 16.2) as a way of generating optimized therapeutic Abs with improved effector functions.
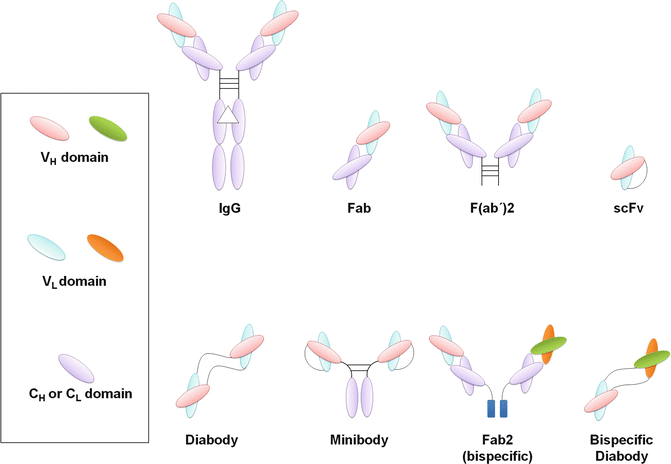
Fig. 16.2
Schematic representation of different antibody fragments with therapeutic applications. Fragment antigen-binding (Fab) and F(ab’)2 may be generated by papain or pepsin digestion of intact IgG, respectively. Other types of antibody fragments can be produced using antibody engineering methods. Single-chain fragment variables (scF vs) are composed of V H –peptide linker–V L (or vice versa). Diabodies are homodimers of scFvs, covalently linked by a short peptide linker. Minibodies consist of two scFv–hinge–CH3 chains covalently connected by disulfide bonds. Bispecific antibodies, in general, consist of variable fragments of two different antibodies. Fab2 and bispecific diabody are two examples of bispecific structures. The triangle on the intact IgG indicates carbohydrates covalently attached to heavy chains
16.6.1 Murine Monoclonal Antibodies
Murine mAbs are entirely derived from mice using hybridoma technology, which involves the fusion of immortalized myeloma cells with B cells from immunized mice [138–142]. However, injection of humans with murine Abs induces the generation of human anti-mouse Abs (HAMA). Not only can these HAMA remove murine Abs upon repeated administrations, but also the formation of antibody-HAMA-complexes has shown to end in mild to severe allergic reactions [143]. Therefore, major shortcomings of intact murine Abs have limited their clinical applications [144].
Although the first mAb approved for clinical applications was a murine IgG2a Ab (OKT3, or muromonab; 1986) [145], many technical efforts were soon made to develop a second-generation mAb appropriate for human administration. Currently, murine Abs serve mainly as radioisotope-labeled agents aiming at targeted killing of tumor cells. Technical advances in recombinant protein engineering, transgenic mice, and phage display has promoted the development of chimeric, humanized and fully human mAbs. This has helped overcome the limitations of intact murine mAbs and resulted in creation of more effective therapeutic agents [146–148].
16.6.2 Chimeric and Humanized Monoclonal Antibodies
The desire to produce murine Abs with less immunogenicity in humans, and more immunologic efficacy, led to the production of chimeric, and humanized mAbs [44, 149, 150]. Chimeric mAbs are produced through grafting the murine variable regions onto human constant regions. Such Abs are 75 % human and much less immunogenic compared to the intact rodent ones, because interspecies immunodominant Ig epitopes are frequently located within the CH2 and CH3 domains of the Fc region [151]. Humanized mAbs, on the other hand, are constructed via engrafting of murine hypervariable Ag binding regions (also named complementarity determining regions (CDRs)) onto human Abs rendering them 85–90 % human, with less immunogenicity than chimeric Abs [151]. It is of note, however, that the binding affinity of the humanized mAbs is often weaker compared to parent murine mAbs. Therefore, additional manipulation needs to be made to humanized Abs to improve their affinity and specificity. These alterations are typically achieved by introducing mutations in the CDRs of Abs [152]. In fact, the majority of currently approved Abs used in oncological applications, and those used in advanced clinical trials are of humanized construct.
16.6.3 Fully Human Monoclonal Antibodies
To further reduce the immunogenicity of chimeric or humanized mAbs, both of which still contain some murine fragments, fully human mAbs were constructed [153, 154]. Replacement of mouse Ig variable and constant domains with those of the human effectively reduces the incidence of anti-antibody response (AAR) hypersensivity reaction [155].
Transgenic mice (bearing human Ig germ line loci) and phage display (the display of Ab fragments on filamentous bacteriophages), as two of the well-established technologies for production of human mAbs, are reviewed here.
16.6.3.1 Human Monoclonal Antibodies from Transgenic Mice
A new approach for development of fully human mAbs is the creation of a mouse strain engineered to produce a large repertoire of human Abs. Such mice are generated by introducing human Ig gene segment loci into the germ lines of mice deficient in Ab production [156]. Interestingly, VDJ recombination and somatic hypermutation of the human germ line Ab genes are carried out in a normal fashion in these mice, thereby producing high-affinity Abs with completely human sequences differing just in glycosylation patterns [157]. Such murine strains may serve as a source of high-affinity human mAbs generated against a broad spectrum of Ags, including those of the human. Panitumumab, ofatumumab and ipilimumab are three fully human anticancer mAbs that have been produced by this technology and approved for use in the clinic (Table 16.2).
Table 16.2
Monoclonal antibodies approved by FDA for cancer therapy
Generic namea | Brand name/company | Targeted antigen | Antibody construct | FDA-approved indication | Approval date |
|---|---|---|---|---|---|
Trastuzumab | HERCEPTIN/Genentech | ERBB2 | Humanized | Breast cancer, metastatic gastric or gastroesophageal junction adenocarcinoma | 1998 |
Bevacizumab | AVASTIN/Genentech and Roche | VEGF | Humanized | Metastatic colorectal cancer, non-squamous non-small cell lung cancer, metastatic breast cancer, glioblastoma, metastatic renal cell carcinoma | 2004 |
Cetuximab | ERBITUX/Bristol-Myers Squibb | EGFR | Chimeric | Head and neck cancer and colorectal cancer | 2004 |
Panitumumab | VECTIBIX/Amgen | EGFR | Human | Metastatic colorectal carcinoma | 2006 |
Ipilimumab | YERVOY/Bristol-Myers Squibb | CTLA-4 | Human | Unresectable or metastatic melanoma | 2011 |
Pertuzumab | PERJETATM/Genentech | ERBB2 | Humanized | Metastatic breast cancer | 2012 |
Conjugated antibodies: solid malignancies | |||||
Ado-trastuzumab emtansine | KADCYLA/Genentech | ERBB2 | Humanized | Metastatic breast cancer | 2013 |
Naked antibodies: hematological malignancies | |||||
Rituximab | Mabthera/Roche, Rituxan/Roche | CD20 | Chimeric | Non-Hodgkin lymphoma, chronic lymphocytic leukemia | 1997 |
Alemtuzumab | Campath/Genzyme | CD52 | Humanized | B-cell chronic lymphocytic leukemia | 2001 |
Ofatumumab | Arzerra/Genmab | CD20 | Human | Chronic lymphocytic leukemia refractory to fludarabine and alemtuzumab | 2009 |
Conjugated antibodies: hematological malignancies | |||||
Brentuximab vedotin | ADCETRIS/Seattle Genetics | CD30 | Chimeric | Refractory Hodgkin lymphoma, systemic anaplastic large cell lymphoma | 2011 |
90Y-labeled ibritumomab tiuxetan | ZEVALIN/IDEC Pharmaceuticals | CD20 | Murine | Relapsed or refractory, low-grade or follicular B-cell non-Hodgkin lymphoma, previously untreated follicular non-Hodgkin lymphoma | 2002 |
Tostitumomab and 131I-labeled tositumomab | Bexxar/GlaxoSmithKline | CD20 | Murine | Rituximab-refractory non-Hodgkin lymphoma | 2003 |
16.6.3.2 Human Monoclonal Antibodies Created Through Phage Display Technology
Phage display was first described by George P. Smith [158] in 1985, when he demonstrated that a foreign DNA fragment can be fused to the gene encoded for pIII coat protein of a filamentous phage and expressed as a fusion protein on the virion surface. A few years later, McCafferty [146] verified that a single-chain fragment variable (scFv) can be presented on a phage surface as a functional protein, while retaining its capability for antigen binding [159]. Today, this is a well-established technology for development of novel fully human Abs. Phage display can mimic the immune system by creating large libraries of Ab genes and selecting for binding to desirable Ags. Depending on the Ab source, there are several types of libraries: immune, naїve, and synthetic libraries. Immunized and naїve phage libraries are constructed through isolating the peripheral lymphocytes from immunized and non-immunized donors, respectively [160]. To create fully synthetic libraries, germ line Ab gene segments, VH, DH, and JH or V κ/λ and J κ/λ are cloned and arranged combinatorially in vitro to reconstitute genes encoding complete VH and VL chains [157]. Although, currently, there is no FDA-approved anticancer therapeutic mAb produced by phage display technology, several of such mAbs are in clinical development [161].
16.6.4 Antibody Fragments
The development of fully humanized Abs was a major breakthrough in therapeutic application of Abs. However, the large size of mAbs together with the presence of the Fc portion may be disadvantageous in some settings since it limits Ab penetration into tumor, especially in the case of solid tumors [162]. In fact, tissue penetration is known as a vital parameter in therapeutic settings, and often severely restricts the complete efficiency of the treatment [41, 163]. In addition, the long half-life of Abs, which is related to their Fc portion, is not appropriate for applications such as radioimmunotherapy or imaging as it may result in irradiation of healthy tissues and high background, respectively [164]. Antibody engineering offered new methods for overcoming these shortcomings which are discussed below.
Antibody fragments including Fab, scFv, diabodies, and minibodies can be produced by elimination of the whole constant region or removal of a part of Fc or its entire portion from Ab [151]. In fact, better renal clearance and improved tumor penetration made such fragments attractive alternatives to the whole Ab molecule for radiotherapy and/or imaging applications [165]. The biodistribution of intact radiolabeled chimeric mAb U36 (125I-cMAB U36) and its radiolabeled-recombinant fragment, 125I-F (ab’) 2, was compared in nude mice bearing head and neck xenograft tumors. Results demonstrated better tumor penetration and superior tumor-to-blood ratio for the latter [164]. Another study demonstrated acceptable tumor uptake of 111In-panitumumab F (ab’) 2 in the athymic mice bearing LS-174T xenografts, suggesting this fragment as a promising candidate for imaging of HER1-positive cancers [166].
scFv fragment (27 kDa) contains the variable domains of one heavy and one light chain linked by a flexible linker and is capable of retaining the binding activity of the full Ig molecule in a monovalent fashion [167]. However, the main disadvantage of scFv is its too short serum half-life (~2 h) compared to the intact Abs (1–2 weeks), which may necessitate a successive administration of the molecule for achieving a proper response [151]. Interestingly, the intracellular expression of anti-Ras neutralizing scFv induced cell death in tumor cells expressing oncogenic Ras [168]. In a preclinical in vitro study, scFv-PEG-lipid conjugate, as an anti-HER2 liposome-inserting agent, was applied to HER2-overexpressing cancer cells [169].
Diabodies are homodimers of scFvs, covalently linked by a short peptide linker of four amino acids [170]. This kind of Ab fragment is a bivalent, medium-size (55 kDa) molecule with a higher avidity and superior tumor retention as compared to a single scFv. Engineered Ab fragments, such as diabodies, and scFv-Fc, have been successfully employed for immuno-positron emission tomography (immunoPET) imaging of cancer cell surface biomarkers in preclinical models [171]. Larger fragments such as minibody (scFv-CH3; 80 kDa) [172] and scFv-Fc (110 kDa) [173] fusion proteins can exhibit even higher tumor uptakes. The longer serum half-life of these species improved their localization and allowed for longer exposure of the target tissue to the Ab fragment. In this regard, genetically engineered minibody and diabody displayed rapid, high-level tumor uptake coupled with rapid clearance from the circulation in the athymic mice bearing LS174T human colon carcinoma [174].
16.6.5 Bispecific Antibodies (BsAbs)
Different modifications have been applied to conventional therapeutic Abs in order to improve their clinical efficacy. Accordingly, bispecific Abs (BsAbs) have been devised that simultaneously target two different Ags on the cell surface [175].
These hybrid proteins can be produced using different approaches such as chemical cross-linking, hybrid of hybridoma (also termed as quadromas) [176], and genetic manipulation (the holes and knob technique) [177]. Today, Ab engineering is capable of producing a wide variety of BsAbs with any antigen-binding combination, and molecular weight, as well as a predictable serum half-life. F (ab’) 2 heterodimer, various types of bivalent and trivalent scFvs, and tetravalent BsAb (including Ab-scFv, dimeric miniantibodies and dimeric antibody-Fc molecules) are some examples of engineered BsAbs in this category [178].
Frequently, BsAbs have been designed to simultaneously bind tumor markers and effector cells. Effector cells such as T-cells are activated via CD3, while others like NK cells, macrophages and neutrophils are generally activated through FcγRIIIa, b and FcγRIIa [179, 180]. In fact, there are many BsAbs with one arm specific to CD3 on cytotoxic T cells and the other arm specific to a tumor Ag such as EGFR [181], HER2 [182], CA-125 [183] or CD20 [184]. Such BsAbs have been administrated in the immunotherapy of NHL, breast, ovarian, and prostate cancers. Blinatumomab, a recombinant bispecific tandem scFv molecule (bispecific T cell engager, BiTE) directed against CD3 and CD19, is undergoing clinical trials and has demonstrated promising results in phase I and II studies in acute lymphoblastic leukemia (ALL) and NHL patients [185, 186]. Although at the beginning of BsAb development, T cells received considerable interest, the attention of recent studies is shifting onto the employment of NK cells. T cells are known as highly motile cells with robust tumor infiltration capacity. However, to become fully activated, these cells needs to interact with co-stimulatory molecules such as B7 on APCs, and this is considered a major drawback to T cell-based modalities [151].
In addition to activation of immune effector cells, BsAbs could be utilized in combination with cytotoxic agents resulting in accumulation of highly active but nonspecific payloads in desired tissues. Recently, recombinant bispecific immunotoxins were produced through fusing a tandem scFv to the catalytic or translocation domain of diphtheria toxin [187–189]. These immunotoxins were directed against CD19 and CD22 and showed improved efficacy against murine xenograft models of B cell malignancies and metastases [187–189].
16.6.6 Antibody Fusion Constructs
Antibody molecules in the fusion constructs are generally used to direct therapeutic agents such as toxins [190], cytokines [191], drugs [192], and radioisotopes [193] to the tumor microenvironment. The rationale behind this approach is the direct and specific delivering of higher concentrations of cytotoxic agents to tumor tissues, while avoiding damage to normal cells [194]. In fact, several potent drugs such as auristatins [195] and maytansinoids [196] (inhibitors of microtubulie assembly) or emtansin [197] (a microtubule polymerization inhibitor) have been utilized in fusion with Abs in cancer therapy. Trastuzumab emtansine is an antibody-drug conjugate consisting of a maytansine derivative (DM1) conjugated to the FDA-approved trastuzumab [198]. Trastuzumab-DM1 has recently been shown to inhibit tumor growth via induction of apoptosis, ADCC and mitotic catastrophe in a trastuzumab/lapaninib (a kinase inhibitor used in breast cancer therapy) resistant murine model, [199].
Aside from drugs, various cytokines (e.g. IL-2, IFN-γ, TNF-α, and GM-CSF) have been investigated as therapeutic agents in conjugation with Abs as explained by their immunomodulatory and antitumor effects. At present, several immunocytokines are undergoing phase I and II clinical trials, and are close to FDA approval [200–202]. One therapeutic approach has combined a humanized Ab recognizing ED-B (extra-domain B of fibronectin) with IL-12 [203]. This conjugated Ab has been evaluated in a phase I study in malignant melanoma and renal cell carcinoma (RCC) patients [203]. Moreover, Ab-IL-2 fusion proteins have been used in several phase I clinical trials to treat melanoma and neuroblastoma [204–206].
Tumor-targeted delivery of radioisotope agents in the form of radioimmunoconjugates is believed to improve its antitumor activity and safety. To minimize toxic effects, the conjugates are commonly designed based upon Abs with short serum half-lives. The only radioimmunotherapy agents licensed by the FDA are yttrium-90 (90Y)-ibritumomab tiuxetan and iodine I 131 tositumomab. Either of these radioimmunoconjugates targets CD20, and each has been associated with potent responses in patients with relapsed NHL, or those with tumors resistant to rituximab [207].
16.6.7 Improvement of Antibody Function
Modifying Abs to improve their function has been a very active area of Ab engineering. Several strategies such as modulating the Fc carbohydrate, and/or protein sequences to enhance immune mediator functions, and altering half-life characteristics are instances of this concept. The existence of oligosaccharides and in particular the N-linked oligosaccharides at Asn-297 in the CH2 domain of IgG1 is crucial for binding to FcγR as well as complement fixation [208–210]. Two independent studies have demonstrated that lack of the fucose moiety from carbohydrate on Asn-297, significantly improves the binding of Ab to FcγRIII and ADCC [211, 212].
Altering protein sequence can be considered as another strategy to improve Ab function. Directed modification of amino acids within the Fc region of Ab leads to alteration of Ab half-life or enhancement of immune-mediated effector functions. A mutated Fc was able to decrease IgG affinity for FcRn, leading to shorter serum half-lives and thus rapid clearance of IgG-toxin or IgG-drug complexes [213]. However, for some therapeutic applications, increasing the half-life is favorable, as it would reduce the need for repetitive injections of the Ab to achieve a therapeutically relevant serum concentration. In one study, utilizing human IgG1 mutants with increased binding affinity to human FcRn led to a 2.5 folds increased serum half-life compared to the wild-type Ab [214].
Monoclonal Abs elicit effector functions following interactions of their Fc portion with various Fc receptors [2]. Hence, increasing the affinity of this interaction by engineering methods can play a major part in the efficacy of Ab-based therapies. Shields et al. determined several amino acids, located on the CH2 domain, as being important in IgG1 binding to FcγR [215]. The binding of IgG1 to FcγRIIIa, the major receptor mediating ADCC by NK cells, was 51 % higher when alanine mutations were made at Ser298, Glu333 and Lys334. Notably, this mutant resulted in greater NK-mediated ADCC compared to a higher concentration of native IgG1 [215].
16.7 Evaluation of Antibody Efficacy
16.7.1 Preclinical Evaluations
Preclinical evaluation of Abs aims at predicting their potential pharmacologic and toxicologic effects in humans.
Different kinds of antitumor activities are evaluated by in vitro tests including inhibition of growth (e.g. trastuzumab [216, 217]), inhibition of metastasis or angiogenesis (e.g. bevacizumab [218, 219]), induction of apoptosis (e.g. rituximab [220, 221]), and induction of secondary immune functions such as ADCC (e.g. trastuzumab [216, 217] or CDC (e.g. rituximab) [220].
The in vivo preclinical studies, on the other hand, can provide valuable information about product-specific dose level, dosing regimen, route of delivery, treatment duration, pharmacokinetics, pharmacodynamics, toxicity [222, 223], and sensitization to chemotherapy [224] or radiotherapy [225].
Choosing the most relevant animal model is a critical step for successful preclinical safety evaluation of a mAb [226–228]. The species- and target-specific nature of mAbs often rules out the use of rodents and in some cases makes it difficult to find the appropriate species. A non-human primate, if ethically justified, could be regarded as the species of choice for human/humanized mAbs [222]. To achieve a thorough assessment, some prefer to use different models including mouse, rat, and monkey as in a study of humanized-anti CD40 mAb (SGN-40) [229].
16.7.2 Clinical Evaluations
Valuable information on the whole procedure of clinical safety evaluation of mAbs have been provided by various regulatory agencies. In 1997, FDA released a revised version of “Points to Consider (PTC) in the Manufacturing and Testing of Monoclonal Antibody Products for Human Use”. This document presents a useful guideline for designing a clinical safety evaluation program of mAbs in areas such as dose estimation, pharmacokinetic evaluation and immunogenicity consideration [223].
A critical step in the clinical evaluation of a therapeutic mAb is to assess its biodistribution, which is the ratio of Ab access to the tumor vs. normal tissues [130, 230, 231]. This step is essential for predicting Ab toxicity [231, 232], defining an appropriate Ab dose regimen, and determining the potential impacts of Ag saturation when using high Ab doses. Scott et al. used a model of a clinical trial that incorporated biodistribution, pharmacokinetic and pharmacodynamic evaluations with toxicity assessment [230] to the first-in-human clinical trials of several anticancer Abs [130, 230, 231, 233]. Further pharmacodynamic assessment methods, such as computerized tomography with magnetic resonance imaging, plasma-based protein, cell and genomic analyses, and tumor biopsies can also be used to evaluate the clinical efficacy of newly designed mAbs [234].
16.8 Clinically-Approved Monoclonal Antibodies
At the beginning of the twentieth century, Paul Ehrlich postulated “magic bullet” as a tool for specific targeting of diseases [235]. His hypothesis became practical with the development of an efficient method for generation of mAbs, in 1975, by Kohler and Milstein [138]. Since then, these molecules have been known as ideal tools for therapy and imaging applications [151]. In this regard, mAb-based therapy of cancer has been used as a new therapeutic modality that has rapidly been adapted in many cancer types [236] and also received a great deal of interest by pharmaceutical companies. This interest has partly been stimulated due to the well-defined safety, efficacy and quality of mAbs, and also because physicians and patients have clearly accepted mAbs as innovative therapeutics [153].
In 1982, for the first time, a therapeutic mAb was successfully used to treat B-cell lymphoma patients [237]. Consequently, Ehrlich’s magic bullet hit the target by introducing rituximab (1997) and trastuzumab (1998) as the first chimeric and humanized FDA-approved mAbs for cancer therapy, respectively [235]. Since 1997, 13 mAbs including seven mAbs specific to solid tumors and six mAbs specific to hematological malignancies have received FDA approval (Table 16.1). Here, we provide an overview of trastuzumab, and bevacizumab (applied for solid tumors) and rituximab (applied for hematological malignancies) as instances of the most successful therapeutic mAbs in clinical oncology [2].
16.8.1 Trastuzumab
Overexpression of human epidermal growth factor receptor-2 (HER2, c-erbB-2/neu, HER2/neu) is reported in approximately 15–20 % of human breast cancers and is associated with a more aggressive disease and poor disease-free survival [238–240]. Trastuzumab is a recombinant humanized mAb (rhumAb 4D5) reacting with an extracellular region of HER2 protein and inhibiting growth of the breast cancer cell line, SKBR-3 [241]. In a pivotal phase III clinical trial on metastatic breast cancer (MBC) patients with HER2 amplification, addition of trastuzumab to the chemotherapy regimen was associated with a few months delay in disease progression (median, 7.4 vs. 4.6 months), a higher rate of objective response (50 % vs. 32 %), and a longer duration of response (median, 9.1 vs. 6.1 months) and survival (median, 25.1 vs. 20.3 months) [242]. Subsequently, four major international studies corroborated that trastuzumab either following or in combination with chemotherapy could reduce the risk of relapse and death by approximately 50 and 33 %, respectively, in HER2-positive early breast cancer patients [243].
Although trastuzumab is accepted as the standard drug in the breast cancer therapy, its use has commonly led to favorable results in a small portion of human breast cancers [238–240]. In addition, up to 40 % of patients with MBC do not respond to trastuzumab-based regimens and in those who respond, the median progression time is less than 1 year [244, 245]. Moreover, acquired trastuzumab resistance is a serious concern ending in disease progression [245, 246]. Notably, due to HER2 expression on cardiomyocytes, cardiac toxicity issues such as symptomatic congestive heart failure has been observed in some of the patients receiving trastuzumab therapies [247, 248]. In general, these shortcomings call for creation of novel and improved Ab-mediated therapies for MBC. Pertuzumab has recently been approved by FDA as a new humanized mAb that blocks HER2 dimerization [244]. This mAb in combination with trastuzumab, and docetaxel is a standard of care for patients with previously untreated MBC [249].
16.8.2 Bevacizumab
As mentioned earlier, vascular endothelial growth factor (VEGF) is a proangiogenic molecule with a critical role in tumor metastasis [250]. Bevacizumab is a humanized mAb that inhibits VEGF activity and is mainly used in combination with chemotherapy for treatment of many types of advanced cancers such as colorectal cancer, RCC, NCLCs, ovarian cancer and glioblastoma [251–257]. The addition of bevacizumab to cytotoxic chemotherapy has improved response rates and survival of patients with metastatic colorectal cancer (mCRC) [258]. Moreover, in a phase III trial, the increase in overall survival of mCRC patients attributable to bevacizumab was 4.7 and 2.1 months following first-line and second-line therapies, respectively [259, 260]. Bevacizumab-based therapy resulted in improved clinical responses in other malignancies as well. For instance, incorporation of bevacizumab to a chemotherapy regimen produced a 2 months clinically relevant improvement in overall survival in NSCLCs compared to chemotherapy alone [256].
Regardless of the utility of several FDA-approved mAbs for cancer treatment, the therapeutic application of mAbs for solid tumors encounters several problems, which are discussed in Sect. 16.11. Compared with solid tumors, targeting of hematological malignancies has proven less complicated because mAbs have easy access to malignant cells allowing for administration of lower Ab doses to achieve potent therapeutic results. Here, rituximab is addressed as the first mAb approved for the treatment of hematological malignancies.
16.8.3 Rituximab
Rituximab is a chimeric mAb specific to CD20, the first Ag targeted for therapeutic purposes and expressed by more than 90 % of B-cell lymphomas [261]. Randomized studies have demonstrated that rituximab induces reasonable antitumor responses in patients with various lymphoid malignancies of B-cell origin, including indolent (e.g. follicular lymphoma (FL)) and aggressive (e.g. diffuse large B-cell lymphoma (DLBC)) forms of NHL (NHL), and CLL. Non-comparative studies have also shown an activity in all other lymphomas [261–263].
A multicenter phase II study on relapsed low grade FL patients showed an overall remission rate of 48 %, (including 6 % of complete response (CR)), and a median progression time of 13 months following rituximab therapy [264]. In untreated FL patients, utilization of rituximab as the first-line therapy along with maintenance therapies led to the improvement of the overall response rate from 47 % (7 % CR) after initial treatment to 73 % (37 % CR) following maintenance treatment [265]. Consolidation therapy with 90Y-ibritumomab tiuxetan, which targets CD20, in the first remission of advanced-stage FL, increased the 8-year overall progression-free survival rate from 22 to 41 %. Interestingly, the median time for the next treatment step was 8.1 years for 90Y-ibritumomab vs. 3.0 years for control [266].
Furthermore, utilization of rituximab in combination with fludarabine and cyclophosphamide led to a significant improvement of the overall survival in CLL patients. Consistently, single-agent rituximab was efficient, even in patients with treatment-refractory or poor-prognosis CLL so that the overall response rate was 90.9 % with a complete remission rate of 63.6 %. Moreover, the median progression-free survival was 28.5 months, and the median duration of response was 26 months [267]. Nonetheless, administration of rituximab as a single agent to CLL has limited clinical activity inasmuch as it generally does not eradicate leukemia from the marrow. However, when employed in combination with chemotherapy, rituximab can improve the survival of patients relative to that of those treated with chemotherapy alone. Subsequently, FDA approved the use of rituximab in combination with fludarabine monophosphate and cyclophosphamide in previously untreated and chemotherapy-treated CD20+ CLL [268].
16.8.4 Therapeutic Monoclonal Antibodies Approved by Non-FDA Organizations
Apart from those authorized by FDA, there are mAbs that are approved outside the United States for cancer therapy (e.g. catumaxomab and nimotuzumab) [269, 270]. For instance, catumaxomab, a trifunctional Ab specific to epithelial cell adhesion molecule (EpCAM) on tumor cells, CD3 on T cells, and Fcγ receptors on accessory cells was approved by the European Union for the treatment of patients with malignant ascites generated by EpCAM-positive carcinomas [271]. Moreover, nimotuzumab, a humanized mAb against EGFR, was developed in Cuba and is approved to treat patients with head and neck cancer, glioma, and nasopharyngeal cancer in more than 20 countries in Asia, South America, and Africa [269, 270, 272].
16.9 Monoclonal Antibodies Currently Undergoing Clinical Trials
The current research is mainly focused on innovative mAbs to novel targets in order to overcome the current limitations of mAb therapy. At present, there are around 350 mAbs with potential applications for a wide range of diseases. Historically, about 50 % of these Abs recognize tumor Ags [273]. Although most of these mAbs are in initial development stages, more than one hundred anticancer mAbs are being evaluated in different phases of clinical trials [274]. Hence, in near future the number of approved mAbs is expected to rise significantly, which could help to improve the outcome of cancer patients by overcoming the current therapeutic limitations. This section briefly introduces some antitumor mAbs that are currently undergoing clinical trials. Several of the mAbs in trials try to provide an opportunity for treatment of untreatable cancers through targeting of novel tumor Ags. For instance, intetumumab, a humanized mAb against human αV integrin, has been successfully tested in phase I/II clinical trials as the first-line treatment in patients with metastatic castration-resistant prostate cancer [275, 276].
Some innovative mAbs, target the well-validated Ags that were previously targeted with the approved mAbs, such as necitumumab (a fully human IgG1, passed phase I of clinical trial in advanced solid malignancies); and nimotuzumab (a humanized IgG1, passed phase I of clinical trial in NSCLC), which both bind specifically to EGFR [277–279]. Some newly designed mAbs in this category are those attempting to improve the functionality of previously-approved mAbs. For instance, obinutuzumab (GA-101), a glycoengineered humanized mAb, binds with high affinity to CD20 type II epitope, resulting in the induction of much stronger ADCC and superior cell killing properties compared to rituximab [280, 281]. Moreover, a phase I/II clinical trial demonstrated that GA-101 has a similar safety profile comparable to that of rituximab, and exhibits promising efficacy in patients with relapsed/refractory CD20-positive lymphoid malignancies [281–283].
Furthermore, there are mAbs designed to bridge cancer and immune cells. A BsAb, named blinatumomab, with dual specificity for CD19 and CD3, potentially engaged cytotoxic T cells for redirected lysis of tumor cells [284]. Consistently, blinatumomab therapy led to a higher degree of in vitro lysis of human lymphoma cells, and was efficient at much lower concentrations compared to rituximab [285]. A phase II trial indicated that blinatumomab could induce complete long-lasting remission in B-lineage ALL patients with persistent or relapsed minimal residual disease (MRD). According to the results, blinatumomab administration induced a 76 % MRD response rate defined as MRD negativity within four cycles of treatment [185, 286].
Finally, immunotoxins are another class of mAbs under clinical investigation. Moxetumomab pasudotox, which is a recombinant immunotoxin composed of the Fv fragment of an anti-CD22 mAb fused to a 38-kDa fragment of Pseudomonas exotoxin A, passed phase I clinical trial with safety and activity in relapsed/refractory hairy cell leukemia (HCL) [287]. Furthermore, this mAb is being evaluated in phase I trials in patients with CLL, B-cell lymphomas, and childhood ALL [288].
16.10 Combinational Monoclonal Antibody-Based Modalities
A brief review of the so far published data on cancer therapy reveals that a single method, such as Ab-based therapy, per se would not be efficacious enough to eradicate the fully armed tumor cells. Hence, in recent years researchers have employed multimodality approaches, which utilize more than a single antitumor agent [120, 289, 290]. This section describes the studies that have examined the effectiveness of combining Ab-targeting with additional common antitumor strategies.
16.10.1 Combination with Chemotherapy
Chemotherapy is one of the methods widely used in combination with Ab therapies to treat various cancers. This method is known to support antitumor immune responses via inducing tumor cell death, eliminating Tregs, and/or making tumor cells more sensitive to lysis by CTLs. Ab-targeted strategies, on the other hand, are believed to render tumor cells more susceptible to chemotherapeutic drugs [291, 292]. An anti-EGFR mAb in combination with chemotherapy could improve overall and/or progression-free survival compared to each agent alone, in patients with mCRC [293]. Moreover, the combination of AZD8055, a rapamycin analogue, and a CD40 agonist mAb, was employed to treat a murine model of metastatic RCC. Notably, the mixture provoked a robust antitumor response in terms of increased infiltration, stimulation, and proliferation of NK cells and CD8+ T cells in metastatic areas compared with what was observed following the use of each treatment alone [294].
Nevertheless, to achieve potent antitumor results one must take into account the probable factors affecting each of the strategies used in a combination therapy approach. For instance, although generally effective, anti-EGFR mAb combined with chemotherapy would be of no therapeutic value if used to treat patients bearing KRAS mutant tumors [293, 295].
16.10.2 Combination with Radiotherapy
Radiotherapy, similar to chemotherapy, has extensively been used in combination with antitumor Abs. The traditional perception of radiotherapy function as a cytocidal weapon decreasing tumor metastasis has recently been shifted to that of a potent adjuvant helping immunotherapy. In fact, current evidence suggests that ionizing radiation per se can successfully induce immunogenic cell death leading to effective activation of antitumor immune responses [296, 297]. However, it should be noted that induction of a potent immunogenic cell death depends upon each tumor’s intrinsic features as well as the genetic polymorphism for certain genes in each host [298, 299].
Additional proimmunogenic mechanisms have been shown to be promoted by ionizing radiation. For instance, chemokines including CXCXL9 and CXCL10, involved in T cell recruitment, were released following radiotherapy of different tumors [300–302]. Interleukin 1β and TNF-α are examples of proinflammatory cytokines induced by radiation [300, 303, 304]. Moreover, sublethal doses of radiation has been shown to enhance the expression of certain molecules on tumor cells rendering them more susceptible to recognition and killing by tumor-specific T cells [297]. On the other hand, radiation therapy has been reported to induce several immunosuppressive mechanisms instead of immune stimulation. There is evidence that radiation activates the latent form of TGF-β, an immunomodulatory cytokine involved in tumor progression [305, 306]. Moreover, radiotherapy has been indicated to induce tolerogenic properties in macrophages [307, 308]. Furthermore, an increase in the number of Tregs has been reported in some patients receiving radiation as an anti-tumor modality [309, 310].
Hence, radiation has the capacity to induce either proimmunogenic or immunosuppressive responses. In most cases, favorable impacts of radiotherapy dominate over the unfavorable ones. However, this is insufficient to thoroughly shift the balance of immune responses against tumor cells in the absence of accompanying immunotherapies [297].
In fact, promising results have been obtained by several preclinical studies that have combined radiotherapy with Ab targeting. Antibody blockade of CTLA-4 combined with local radiation in a murine model of breast cancer significantly increased the survival rate due to the induction of effective T cell responses, whereas radiotherapy alone could only delay tumor growth, and anti-CTLA-4 mAb by itself was completely ineffective [311]. Consistently, the metastasis of poorly immunogenic colorectal and mammary carcinomas was successfully inhibited by a combination of radiation and anti-CTLA-4 mAb in mice [312]. Antibody targeting of 4-1BB (CD137), a critical receptor on T cell surface, combined with ionizing radiation has resulted in several other beneficial antitumor effects [313–315]. Interestingly, the combination of radiotherapy and anti-CTLA-4 Ab has also led to promising results in clinical trials [316]. In a case report of melanoma, treatment of the patient with ipilimumab (anti-CTLA-4 Ab) following radiation [317] could mimic the successful results previously observed in murine models [311, 312].
Nonetheless, to exploit the full potential of this type of combination to treat cancers entails the establishment of standard radiation regimens, which can result in effective domination of proimmunogenic over immunosuppressive responses. To this end, investigators are recommended to test different doses and frequencies of radiation in combination with each immunotherapeutic method for every cancer type and choose the optimal combination strategy [297, 312, 318].
16.10.3 Combination with Other Immunotherapeutic Methods
Antibody-based therapeutic methods have also been used together with other immunotherapeutic strategies to outsmart tumor-associated evasion mechanisms. For instance, anti-4-1BB mAb, as a CD4+ T cell adjuvant, was applied together with in vitro activated antitumor T cells to a murine model of microscopic pulmonary metastasis. The combination was advantageous over Ab administration or adoptive T cell therapy alone. In fact, anti-4-1BB mAb served as an efficacious adjuvant through augmenting the anti-tumor function of transferred T cells and resulted in persistence of infiltrated effector T cells [319]. However, one major disadvantage of using anti-4-1BB mAb is its toxic effects in higher doses. To overcome this issue, one study employed a combination of lower doses of anti-4-1BB and tumor lysate-pulsed DCs for treatment of liver metastatic colon cancer. This nontoxic combination strategy resulted in a significant increase in tumor rejection comparable to the level obtained with higher toxic doses of anti-4-1BB alone [320]. In a very recent study, T cells, engineered to express a type of tumor-specific MUC-1 receptor, were adoptively used to target prostate cancer cells. However, the vaccine efficacy was hindered by the heterogeneous expression of MUC-1 by tumor cells. Interestingly, the addition of a type of conventional anti-androgen mAb to the treatment regimen, could improve the antitumor effects in vitro [321]. These examples substantiate the advantage of employing alternative immunotherapeutic approaches along with Ab-based modalities to obtain more potent and less toxic antitumor responses.
16.10.4 Other Combinational Approaches
In addition to the aforementioned more popular combination approaches, researchers have examined the efficacy of employing several less-known modalities. For instance, a combination of Abs against two growth factors, secreted by human pancreatic cell lines, was successfully used to improve the efficacy of chemotherapy in pancreatic cancer patients [322]. Moreover, in a recent murine model of breast cancer, a recombinant protein with the capacity to bind to epithelial cell junctions was used as a partnering treatment for anti-EGFR-mAb. Interestingly, the cell junction opener protein could improve the intratumoral penetration of mAb culminating in robust antitumor responses [323].
Overall, with regard to Ab-based antitumor strategies, data obtained from preclinical and clinical studies corroborate that combinatorial approaches are undoubtedly superior to simple utilization of a mAb alone. Designing the most efficacious approaches entails gaining a precise understanding of the cellular and molecular events underlying the interaction between the combined methods. Notably, the mAb of interest needs to be used in combination with a range of successful immunostimulating methods to choose the best partnering agent.
16.11 Current Limitations in Monoclonal Antibody-Based Therapies
16.11.1 Tumor Escape
It often occurs that patients with the same cancer type respond differently to a certain Ab-based strategy. This could be in part attributed to the diverse mechanisms tumor cells use to escape immune responses [324]. Here, we describe major mechanisms underlying tumor resistance to Ab-based modalities.
One reason for the resistance to mAb therapy in most cancer patients might be the presence of agents that inhibit CDC [325]. Protectin (CD59) inhibits homologous CDC by preventing formation of the membrane attack complex, thereby inhibiting cell lysis [326]. In fact, a great deal of evidence indicates that CD59 is highly effective in protecting NHL, melanoma and CLL cells from antibody-mediated CDC and up-regulation of CD59 is an important determinant of sensitivity to Ab treatment in such cancers [327, 328].
Tumor cells might circumvent ADCC via expression of NK cell inhibitory molecules such as HLA-G, a non-classical HLA class I [329], which is known to be expressed on melanoma and other malignancies [330–332]. Interestingly, rituximab-mediated NK cell lysis depends on the HLA class I expression level on B-lymphoma cells [329].
To evade Ab-mediated therapies, tumor cells can downregulate the expression of Ags targeted by mAbs. Intriguingly, high receptor expression is known to be associated with a favorable response to trastuzumab. However, due to target receptor downregualtion following Ab therapy, a proper response may not always be achieved [2]. Similarly, acquired rituximab resistance in B-cell lymphomas following exposure to rituximab has been associated with reduced levels of CD20 [333–335].
Masking of target proteins on tumor cells is another tumor escape mechanism. Resistance to trastuzumab was associated with increased expression of the membrane-associated glycoprotein MUC-4, which was shown to bind and sterically prevent HER2 from binding to trastuzumab [336–338].
Tumor resistance to Ab targeting might occur because of the induction of compensatory or alternative signaling by other cell surface receptors. Cetuximab (anti-EGFR mAb)-resistant tumors have been shown to escape Ab treatment through increased expression of G-protein coupled receptors [324, 339]. Furthermore, resistance to cetuximab treatment in colorectal cancers is often related to point mutations of KRAS and its downstream signaling molecules (e.g. BRAF) [340–343].
16.11.2 Relatively Low Single Agent Activity
Although numerous therapeutic mAbs have been approved for clinical use, in most cases, the overall response to a single mAb remains low. Accordingly, mAbs are commonly used in combination with other treatment modalities to achieve more favorable results (discussed in Sect. 16.10).
16.11.3 Low Tissue Penetration
Molecular size plays a key role in tumor penetration of therapeutic mAbs, and in fact, the diffusion rate inversely correlates with the cube root of molecular weight. Therefore, mAbs, as large molecules, would have difficulty diffusing into solid tumors, resulting in increased resistance of larger tumors to mAb-based modalities [344].
Using mAbs with high affinity can further diminish tumor penetration of Abs, a factor called “binding site barrier effect” [345]. In fact, there are several reports verifying that very high affinities can lead to suboptimal antitumor responses [346, 347]. The tight binding of mAbs to their Ag targets on the outer surface of solid tumors hampers their deeper penetration into tumor mass. Therefore, development of mAbs with optimal affinities for tumor Ags would result in efficient antitumor responses. However, achieving robust clinical responses mandates the consideration of several factors including Ag density, internalization, association and dissociation rates, therefore, it is not always easy to develop perfect mAbs.
16.11.4 Fc-Fc Receptor Interactions and Associated Limitations
Elimination of tumors using mAbs that promote ADCC meets several challenges. First of all, a successful ADCC process requires a high affinity between Fc of a mAb and its receptor on effector cells; this is a major problem since a high percentage of the population express low affinity variants of the Fc receptor [112]. It has been shown that the presence of a valine (V) at position 158 of FcγRIIIa/CD16a instead of a phenylalanine (F) improves the FcR affinity for IgG [348, 349], and this replacement is shown to correlate with improved responses to rituximab therapy [112, 350].
Secondly, the glycosylation pattern of the Fc fragment of a mAb can be of major importance when working with therapeutic mAbs. In particular, the CH2 domain of IgG1 is glycosylated (Asn-297) and this has been shown to have a key role in modulating the interaction of Fc with FcγRIIIa, thereby affecting the Ab efficacy. More specifically, the presence of fucose residues in the carbohydrate moiety has been reported to end in decreased ADCC efficiency [212].
Related posts:
 Novel Strategy of Cancer Immunotherapy: Spiraling Up
Novel Strategy of Cancer Immunotherapy: Spiraling Up
 Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
 Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response
 Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


