Antibodies are produced by B cells and arise in response to exposures to a variety of structures, termed antigens, as a result of a series of recombinations of V, D, and J germline genes. Immunoglobulin-G (IgG) molecules are most commonly employed as the working backbones of current therapeutic monoclonal antibodies, although various other isotypes of antibodies have specialized functions (e.g., IgA molecules play important roles in mucosal immunity, IgE molecules are involved in anaphylaxis). The advent of hybridoma technology by Kohler and Milstein5 made it possible to produce large quantities of antibodies with high purity and monospecificity for a single binding region (epitope) on an antigen.
The mechanisms that antibody-based therapeutics employ to elicit antitumor effects include focusing components of the patient’s immune system to attack tumor cells6,7 and methods to alter signal transduction pathways that drive tumor progression.8,9 Antibody-based conjugates employ the targeting specificity of antibodies to deliver toxic compounds, such as chemotherapeutics, specifically to the tumor sites.
Structural and Functional Domains
An IgG molecule is typically divided into three domains consisting of two identical antigen-binding (Fab) domains connected to an effector or Fc domain by a flexible hinge sequence. Figure 29.1 shows the structure of an IgG molecule. IgG antibodies are comprised of two identical light chains and two identical heavy chains, with the chains joined by disulfide bonds, resulting in a bilaterally symmetrical complex. The Fab domains mediate the binding of IgG molecules to their cognate antigens and are composed of an intact light chain and half of a heavy chain. Each chain in the Fab domain is further divided into variable and constant regions, with the variable region containing hypervariable, or complementarity determining regions (CDR) in which the antigen-contact residues reside. The light and heavy chain variable regions each contain three CDRs (CDR1, CDR2, and CDR3). All six CDRs form the antigen-binding pocket and are collectively defined in immunologic terms as the idiotype of the antibody. In the majority of cases, the variable heavy chain CDR3 plays a dominant role in binding.10
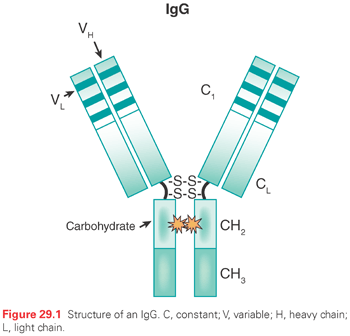
The different isotypes of immunoglobulins are defined by the structure and function of their Fc domains. The Fc domain, composed of the CH2 and CH3 regions of the antibody’s heavy chains, is the critical determinant of how an antibody mediates effector functions, transports across cellular barriers, and persists in circulation.7,11
MODIFIED ANTIBODY-BASED MOLECULES
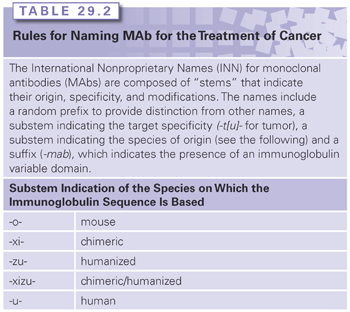
Advances in antibody engineering and molecular biology have facilitated the development of many novel antibody-based structures with unique physical and pharmacokinetic properties (see Fig. 29.1). These include chimeric human-murine antibodies with human-constant regions and murine-variable regions,12 humanized antibodies in which murine CDR sequences have been grafted into human IgG molecules, and entirely human antibodies derived from human hybridomas and, more recently, from transgenic mice expressing human immunoglobulin genes.13 An accepted naming scheme based on “stems” was developed by the World Health Organization’s International Nonproprietary Names (INN) for pharmaceuticals and is employed in the United States (Table 29.2). Engineering has also facilitated the development of antibody-based fragments. In addition to the classic, enzymatically derived Fab and F(ab′)2 molecules, a plethora of promising IgG-derivatives have been developed that retain antigen-binding properties of intact antibodies (see Fig. 29.1; for review see Robinson et al.14). The basic building block for these molecules is the 25 kDa, monovalent single-chain Fv (scFv) that is comprised of the variable domains (VH and VL) of an antibody fused together with a short peptide linker. Novel, bispecific antibody-based structures can facilitate binding to two tumor antigens or bridge tumor cells with immune effector cells to focus antibody-dependent cell-mediated cytotoxicity (ADCC) or killing by T cells. An example of the former is MM-111, a bispecific gene-fused molecule composed of an anti-HER2 scFv connected to an anti-HER3 scFv via a modified form of human serum albumin.15 Examples of the latter mechanism include small scFv-based bispecific T-cell engagers (BiTE) such as the anti-CD3/anti-CD19 molecule blinatumomab16 and larger MAb-based antibodies such as catumaxomab, a rat/mouse anti-CD3/EpCAM bispecific MAb produced via quadroma technology.17 Both classes of bispecifics endow selectivity and targeting properties that are not obtainable with natural antibody formats.
FACTORS REGULATING ANTIBODY-BASED TUMOR TARGETING
Antibody Size
Nonuniform distribution of systemically administered antibody is generally observed in biopsied specimens of solid tumors. Heterogeneous tumor blood supply limits uniform antibody delivery to tumors, and elevated interstitial pressures in the center of tumors oppose inward diffusion.18 This high interstitial pressure slows the diffusion of molecules from their vascular extravasation site in a size-dependent manner.19,20 The relatively large transport distances in the tumor interstitium also substantially increase the time required for large IgG macromolecules to reach target cells.21
Tumor Antigens
Access to the target antigen is undoubtedly a critical determinant of therapeutic effect of antibody-based applications. Such access is regulated by the heterogeneity of antigen expression by tumor cells. Shed antigen in the serum, tumor microenvironment, or both may saturate the antibody’s binding sites and prevent binding to the cell surface. Alternatively, a rapid internalization of an antibody/antigen complex, although critical for antibody–drug conjugates (ADC), may deplete the quantity of cell surface MAb capable of initiating ADCC or cytotoxic signal transduction events. Finally, target antigens are normally tumor associated rather than tumor specific. Tumor-specific antigens are both highly desirable and rare. Typically, such antigens arise as a result of unique tumor-based genetic recombinations, such as clonal immunoglobulin idiotypes expressed on the surface of B-cell lymphomas.22
Antibody affinity for its target antigen has complex effects on tumor targeting. The binding-site barrier hypothesis postulates that antibodies with extremely high affinity for target antigen would bind irreversibly to the first antigen encountered upon entering the tumor, which would limit the diffusion of the antibody into the tumor and accumulate instead in regions surrounding the tumor vasculature.23,24 Similarly, in tumor spheroids, the in vitro penetration of engineered antibodies is primarily limited by internalization and degradation.25 The valence of an antibody molecule can increase the functional affinity of the antibody through an avidity effect.26–28
Half-Life/Clearance Rate
The concentration of intact IgG in mammalian serum is maintained at constant levels with half-lives of IgGs measured in days. This homeostasis is regulated in part by the major histocompatibility complex (MHC)-class I–related Fc receptor, FcRn (n = neonatal), a saturable, pH-dependent salvage mechanism that regulates quality and quantity of IgG in serum. This mechanism can be exploited via mutations in the Fc portion of an IgG to modulate IgGs pharmacokinetics.29,30 Indeed, multiple strategies have been developed to increase the serum persistence of antibody-based fragments and other classes of protein therapeutics.14,31
Glycosylation
IgGs undergo N-linked glycosylation at the conserved Asn residue at position 297 within the CH2 domain of the constant region. Glycosylation status of the residue has long been known to impact the ability of IgGs to bind effector ligands such as FcγR and C1q, which, in turn, affects their ability to participate in Fc-mediated functions such as ADCC and complement-dependent cytotoxicity (CDC).32–34 The glycosylation of MAbs can be altered to increase ADCC by producing them in a cell line engineered to express β(1,4)-N-acetylglucosaminyltransferase III (GnTIII), the enzyme required to add the bisecting GlcNAc residues.33 Defucosylation of antibody Fc domains is also associated with enhanced ADCC, and in a recently completed multicenter phase II trial of a defucosylated anti-CC chemokine receptor 4 (CCR4), MAb was associated with meaningful antitumor activity, including complete responses and enhanced progression-free survival (PFS).35
The majority of monoclonal antibodies approved for clinical use display intrinsic antitumor effects that are mediated by one or more of the following mechanisms.
Cell-Mediated Cytotoxicity
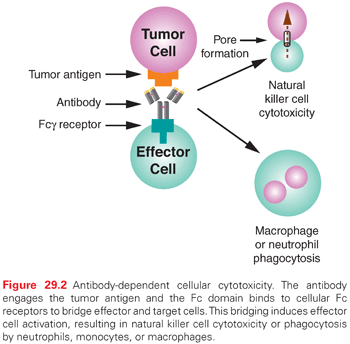
As components of the immune system, effector cells such as natural killer (NK) cells and monocytes/macrophages represent natural lines of defense against oncologically transformed cells. These effector cells express Fcγ receptors (FcγR) on their cell surfaces, which interact with the Fc domain of IgG molecules. This family is comprised of three classes (type I, II, and III) that are further divided into subclasses (IIa/IIb and IIIa/IIIb).36 Recognition of transformed cells by immune effector cells leads to cell-mediated killing through processes such as ADCC and phagocytosis, as shown in Figure 29.2, and can be mediated by FcγRI (CD64), a high affinity receptor capable of binding to monomeric IgG, or FcγRII (CD32) and FcγRIII (CD16), which are low affinity receptors that preferentially bind multimeric complexes of IgG. Signaling through type I, IIa, and IIIa receptors results in the activation of effector cells due to associated immunoreceptor tyrosine-based activation motifs (ITAM), whereas the engagement of type IIb receptors inhibits cell activation through associated immunoreceptor tyrosine-based inhibitory motifs (ITIM).36 Clinical results support the idea that ADCC can play a role in the efficacy of antibody-based therapies. Naturally occurring polymorphisms in FcγRs alter their affinity for human IgG1 and have been linked to clinical response.37,38 A polymorphism in the FCGR3A gene results in either a valine or phenylalanine at position 158 of FcγRIIIa. Human IgG1 binds more strongly to FcγRIIIa-158V than FcγRIIIa-158F, and likewise to NK cells from individuals that are either homozygous for 158F or heterozygous for this polymorphism.39 The FcγRIIIa-158v was a predictor of early response and was associated with improved PFS. A second polymorphism, FcγRIIa-131H/R, did not predict early response but was an independent predictor of time to progression (TTP).38 Taken together, these data suggest that modulating the affinity of MAbs for FcγRIIIa, FcγRIIa, or both may increase the efficacy of therapeutic MAbs.
Each class of FcγR exhibits a characteristic specificity for IgG subclasses.40 Many groups have focused on modifying the Fc domain of IgGs to optimize the engagement of subclasses of FcγR and the induction of ADCC, based on the findings of Shields et al.,29 who performed a series of mutagenesis experiments to map the residues required for IgG1-FcγR interaction. Antibodies such as ocrelizumab, a humanized version of rituximab, have increased binding to low affinity FcγRIIIa variants and are now in clinical trials.
An alternative to modifying the Fc region of MAbs is to create bispecific antibodies (bsAbs) that recognize both a tumor-associated antigen and a trigger antigen present on the surface of an immune effector cell.43 Simultaneous engagement of both antigens can redirect the cytotoxic potential of the effector cell against the tumor.41–43 Such antibodies are capable of eliciting effector function against tumor cell lines in vitro and in animal models. Two HER-2 directed bispecific antibodies, 2B1 and MDX-H210, have been tested in phase I clinical trials.44,45
Bispecific antibodies have a number of distinctive properties, including flexible choices of cytotoxic trigger molecules,46 recruitment of effector function in the presence of excess IgG,42 and custom tailoring of the affinity of the bsAb to match effector cell characteristics. These advantages have been facilitated by improved methods of bsAb production.47 BiTE antibodies represent a novel class of bispecific, single-chain Fv antibodies.48 Promising results have been seen in early phase clinical trials with at least two BiTE antibodies, one of which, blinatumomab, targets CD19/CD3.49 Promising phase I results have also been reported in an interim analysis of an anti-EpCAM/anti-CD3 MT110 BiTE in the setting of advanced lung and gastrointestinal tumors.50
Complement-Dependent Cytotoxicity
In addition to cell-mediated killing (see previous), MAbs can recruit the complement cascade to kill cells via CDC. Although IgM is the most effective isotype for complement activation, it is not widely used in clinical oncology. Similar to ADCC, the human IgG subclass used to construct a therapeutic MAb dictates its ability to elicit CDC; IgG1 is extremely efficient at fixing complement, in contrast to IgG2 and IgG4.51 Antibodies activate complement through the classical pathway, by engaging multiple C1q to trigger activation of a cascade of serum proteases, which kill the antibody-bound cells.52,53 The anti-CD20 MAb rituximab has been found to depend in part on CDC for its in vivo efficacy.54 Antibody engineering approaches have identified residues in the CH2 domain of the Fc region that either suppress or enhance the ability of rituximab to bind C1q and activate CDC.55 The ability to manipulate complement fixation through engineering approaches warrants in vivo testing to determine the impact of these changes on the efficacy and toxicity of MAbs.
Growth factor receptors represent a well-established class of targets for therapeutic intervention. Normal signaling through these receptors often leads to mitogenic and prosurvival responses. Unregulated signaling, as seen in a number of common cancers due to receptor overexpression, promotes tumor cell growth and insensitivity to chemotherapeutic agents. Clinically relevant MAbs can modulate signaling through their target receptors to normalize cell growth rates and sensitize tumor cells to cytotoxic agents. The binding of cetuximab or panitumumab to the epidermal growth factor receptor (EGFR) physically blocks ligand binding56 and prevents the receptor from assuming the extended conformation required for dimerization.57 Pertuzumab binds to the dimerization domain of HER-2, thereby sterically inhibiting subsequent receptor heterodimerization with other ligand-bound family members.58 Alternatively, signaling through growth factor receptors can be indirectly modified by MAbs that bind to activating ligands, as is seen with the anti–vascular endothelial growth factor (VEGF) MAb, bevacizumab.59
MAbs that are not capable of directly eliciting antitumor effects, either by altering signal transduction or directing immune system cells, can still be effective against tumors by delivering cytotoxic payloads. MAbs have been employed to deliver a wide variety of agents, including chemotherapy, toxins, radioisotopes, and cytokines (for review see Adams and Weiner60). In theory, the appropriate combination of toxic agents and MAbs could lead to a synergistic effect. For example, delivery of a therapeutic radioisotope by a MAb would be significantly enhanced if, by binding to its target antigen, the MAb also activated a signaling event that increased the target cell’s sensitivity to ionizing radiation.
Catalytic toxins derived from plants catalytic toxins derived from plants (e.g., ricin) and microorganisms (e.g., Pseudomonas) represent two classes of cytotoxic agent that have been investigated for their utility in immunoconjugate strategies.61 Although there are promising preclinical studies,62 few successful clinical trials have been reported using this approach. In a phase I clinical trial in hairy cell leukemia patients who were resistant to cladribine, 11 of 16 patients exhibited complete remissions with minimal side effects with an anti-CD22 immunotoxin with a truncated form of Pseudomonas exotoxin.63 Clinical trials with other immunotoxins have been associated with unacceptable neurotoxicity64 and life-threatening vascular leak syndrome.65
Immunocytokine fusions have also been investigated as an approach to direct the patient’s immune response to his or her own tumor.66 A number of cytokines have been incorporated into antibody-based constructs, including interleukin-2 (IL-2),67,68 interferon γ (IFN-γ),69 tumor necrosis factor α (TFN-α),69 VEGF,70 and IL-12.71
Antibody–Drug Conjugates
The first ADC, gemtuzumab ozogamicin (Mylotarg), was approved by the FDA in 2000 for the treatment of patients with relapsed CD33-positive acute myeloid leukemia, but was voluntarily withdrawn from the US market by its manufacturer in 2010 after a confirmatory phase III trial (SWOG S0106) recommended, based on results of a planned interim analysis, that Mylotarg randomizations be terminated due to a lack of efficacy in the presence of enhanced toxicity.72 Although two additional randomized trials73,74 suggested that some patient populations may benefit from Mylotarg therapy, the drug remains off the market in the United States.
The majority of ADCs under development employ potent cytotoxic agents that block the polymerization of tubulin (e.g., auristatins or maytansines) or damage DNA (e.g., calicheamicins or pyrrolobenzodiazepines) by employing a variety of linkers and conjugation strategies.75
A variety of ADCs specific for a wide range of oncology targets are currently in clinical evaluation, with the majority of the more advanced agents being tested in the setting of diffuse malignancies.76 The majority of these employ auristatins or maytansines as their payloads. Early observations suggest that cumulative, dose-related peripheral sensory neuropathy can result when auristatins are conjugated to an antibody via a cleavable linker, and dose-limiting thrombocytopenia can result when auristatins and maytansinoids are conjugated to the antibody via an uncleavable linker.76,77
Two ADCs are now approved for use in clinical practice. Ado-trastuzumab emtansine (T-DM1, Kadcyla), an ADC composed of the anti-HER2 MAb trastuzumab linked to DM1,78 is now approved for the treatment of patients with refractory HER2/neu expressing breast cancers. The other, brentuximab vedotin (SGN-35, Adcetris), is an ADC consisting of the anti-CD30 chimeric MAb cAC10 that is linked to three to five molecules of the microtubule-disrupting agent Monomethyl auristatin E. At this point, this drug is approved for use in patients with recurrent systemic anaplastic large cell lymphoma. The clinical data associated with both of these ADCs will be discussed in subsequent sections of this chapter.
Antibodies also can be used to target liposome-encapsulated drugs79 and other cytotoxic agents, such as antisense RNA80 or radionuclides to tumors.
Radioimmunoconjugates
Two anti-CD20 radioimmunoconjugates have been FDA approved for radioimmunotherapy (RIT) of non-Hodgkin lymphoma (NHL). Ibritumomab (Zevalin) and tositumomab (Bexxar) are murine MAbs labeled with yttrium-90 (90Y) and iodine-131 (131I), respectively. Both are associated with impressive clinical efficacy.81,82 Although these radioimmunoconjugates are effective therapeutics, cumbersome logistics surrounding their administration have significantly limited their use. Despite significant preclinical evidence supporting the use of RIT for solid malignancies, clinical results have not demonstrated consistent antitumor activity.60
ANTIBODIES APPROVED FOR USE IN SOLID TUMORS
Trastuzumab
Trastuzumab (Herceptin) is a humanized IgG183 that targets domain IV of the HER2/ErbB2 member of the EGFR/ErbB family of receptor tyrosine kinases. Gene amplification as judged by fluorescence in situ hybridization (FISH) with concomitant overexpression of HER2 protein measured by immunohistochemistry (IHC) is seen in approximately 25% of breast cancers.84,85 HER2 amplification and overexpression is now recognized to also be a critical driver in a subset (7% to 34%) of gastric cancers.86 Trastuzumab inhibits tumor cell growth by binding to HER2 and blocking the unregulated HER2 signaling that is associated with its high level overexpression.
Trastuzumab became the first FDA-approved monoclonal antibody for the treatment of solid tumors based on a series of studies carried out in the setting of HER2-positive metastatic breast cancer.87,88 A subsequent phase III trial investigating trastuzumab in combination with cytotoxic chemotherapy demonstrated an improved response rate compared to chemotherapy alone, from 25.0% to 57.3% with a taxane regimen.89
Trastuzumab is also approved for use in the adjuvant setting based on an approximately 50% reduction in recurrence after 1 year in multiple phase III trials.90–92 Myocardial dysfunction, seen with anthracycline therapy, was observed with increased frequency in patients receiving antibody alone93 or with doxorubicin or epirubicin.
Recognition of HER2 as a driver in a subset of gastric cancers led to an open-label, randomized, phase III trial (ToGA) that investigated the addition of trastuzumab to standard of care chemotherapy94 and showed increased median overall survival with higher levels of HER2 expression. A study by Gomez-Martin et al.95 in 99 patients with metastatic gastric cancer being treated with first-line trastuzumab plus chemotherapy identified a mean HER2/CEP17 ratio of 4.7 to be an optimal cut-off to discriminate between trastuzumab-sensitive and refractory patients.
Pertuzumab
Pertuzumab (Perjeta) is a humanized IgG1 MAb that binds to domain II of HER2 and blocks ligand-dependent dimerization of HER2 with other members of the EGFR family.96 Pertuzumab, in combination with trastuzumab and docetaxel, is approved for use as first-line therapy in HER2-positive metastatic breast cancer patients. Use of the combination is also approved for the treatment of HER2-positive, locally advanced, inflammatory, or high-risk early breast cancer (>2 cm node negative or node positive) in the neoadjuvant setting.
FDA-approval of pertuzumab was based on results of a phase III trial (CLEOPATRA) of 808 patients with locally recurrent, unresectable, or metastatic breast cancer randomized to receive trastuzumab plus docetaxel with or without the addition of pertuzumab. Inclusion of pertuzumab increased the independently assessed PFS by 6.1 months from 12.4 to 18.5 (hazard ratio [HR], 0.62 (95% confidence interval [CI], 0.51, 0.75), p <0.0001], with a trend toward improved overall survival97 that reached statistical significance (p = 0.0008) after an additional year of follow-up.98 The addition of pertuzumab did increase rates of grade 3 adverse events (AE), but it did not adversely affect cardiac function. Accelerated approval was granted for use of pertuzumab in combination with trastuzumab and docetaxel for the neoadjuvant treatment of high-risk early-stage breast cancer. This approval was based on results from a four-arm, open-label phase II study of 417 patients randomized to receive trastuzumab plus docetaxel, pertuzumab plus docetaxel, pertuzumab plus trastuzumab, or the triple combination. The triple combination improved the pathologic complete response (pCR) rate by 17.8% over the trastuzumab plus docetaxel arm (39.3% versus 21.5%) in the pertuzumab arm.99 Follow-up studies to confirm a correlation between pCR and long-term clinical benefit are ongoing.
Cetuximab
Cetuximab (Erbitux) targets the EGFR. This chimeric IgG1 binds to domain III of the EGFR, with roughly a tenfold higher affinity than either EGF or transforming growth factor α (TGF-α) ligands and thereby inhibits ligand-induced activation of this tyrosine kinase receptor. Cetuximab may also function to downregulate EGFR-dependent signaling by stimulating EGFR internalization.100
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


