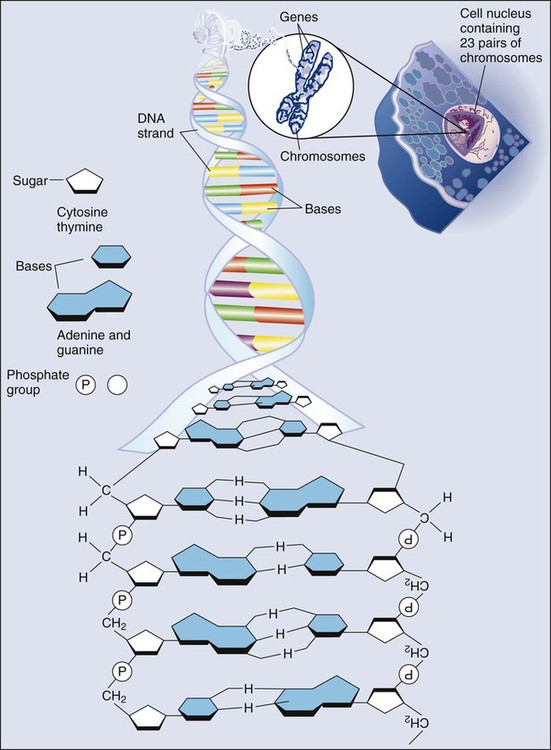
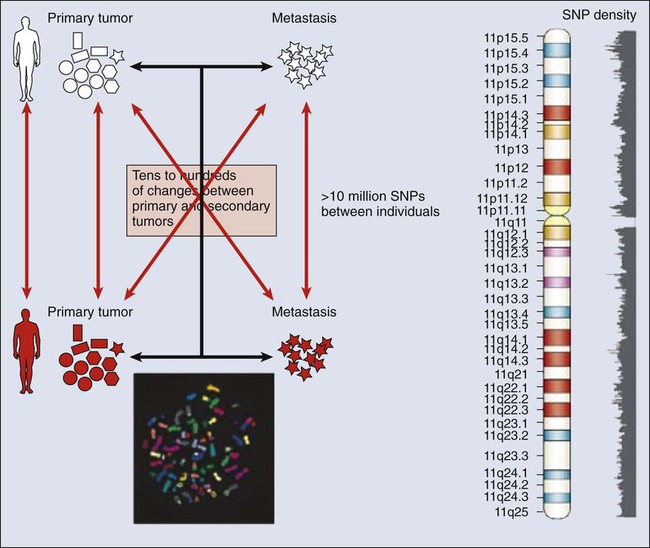
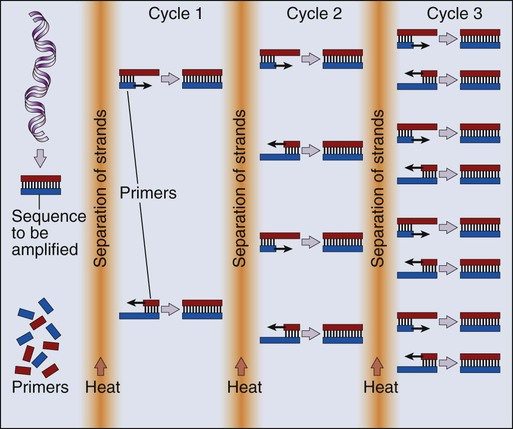
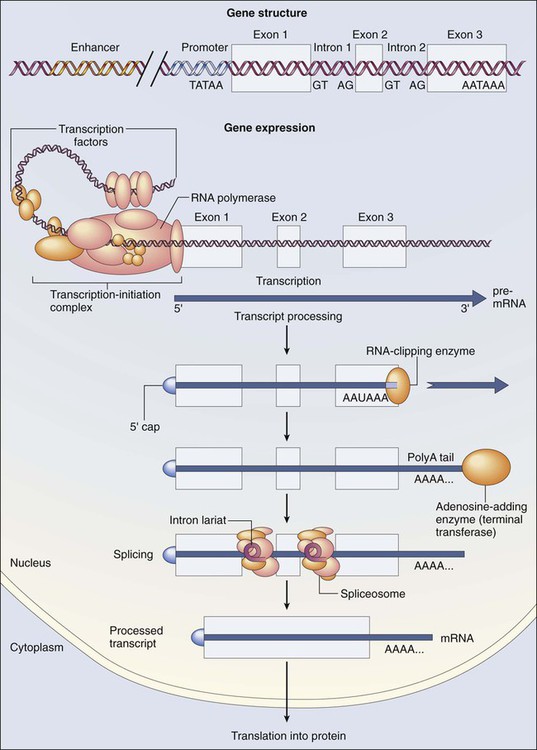
Mauro W. Costa and Nadia Rosenthal • Our understanding and treatment of cancer have always relied heavily on parallel developments in biological research. Molecular biology provides the basic tools to study genes involved with cancer growth patterns and tumor suppression. An advanced understanding of the molecular processes governing cell growth and differentiation has revolutionized the diagnosis and prognosis of malignant disorders. • This introductory chapter relates basic principles of molecular biology to emerging perspectives on the origin and progression of cancer and explains newly developed laboratory techniques, including whole genome analysis, expression profiling, and refined genetic manipulation in animal models, thus providing the conceptual and technical background necessary to grasp the central principles and new methods of current cancer research. All methods for detecting mutations rely on the manipulation of DNA, the basic building block of heredity in the cell. DNA consists of two long strands of polynucleotides that twist around each other clockwise in a double helix (Fig. 1-1). Nucleic acid bases attached to the sugar groups of each strand face each other within the helix, perpendicular to its axis. Only four bases exist: the purines adenine and guanine (A and G) and the pyrimidines cytosine and thymine (C and T). During assembly of the double helix, stable pairings of nucleotides from either strand are made between A and T or between G and C. Each base pair (bp) forms one of the billions of rungs in the long, unbroken ladder of DNA that forms a chromosome. The dramatic increase in genetic complexity conferred by alternate RNA splicing is underscored by the multiple splice patterns of many medically relevant genes, in which different combinations of exons are chosen for the final mRNA transcript, such that one gene can encode many different proteins (Fig. 1-2). The choice of protein isoform to be expressed from a gene with multiple splicing possibilities is a decision that can be perturbed in disease. To date, errors in splicing mechanisms have been associated with a large group of cancers. These errors include mutations in several transcription factors, cell signaling, and membrane proteins. These include the oncogene p53 in more than 12 different types of cancer and mutL homolog 1 protein mutation in hereditary nonpolyposis colorectal cancer. When mutations in the splicing site lead to insertion of novel sequences in the mRNA, the encoded protein can be used as a potential clinical marker, as seen for the transcription factor NSFR in persons with small cell lung cancer. Because of their unique expression in cancer cells, these markers can be further explored as new cancer-specific therapeutic targets. The complete set of DNA sequences carried on all the chromosomes is known as the genome. Although the general map of the genome is shared by all members of a species, the recent sequencing of thousands of individual human genomes has given rise to the new field of genomics, providing us with new tools to reveal the more subtle variations that arise between individuals. These variations are critical, both as a natural engine driving heterogeneity within a species and as a source of predisposition to cancer types. The most common forms of human genetic variations, or alleles, arise as single-nucleotide polymorphisms, or SNPs. Because these allelic dissimilarities are abundant, inherited, and dispersed throughout the genome, SNPs can be used to track racial diversity, personal traits, and susceptibility to common forms of cancer (Fig. 1-3). Our ability to monitor hundreds of thousands of SNPs simultaneously is one of the most important advances in modern medical genetics. Relatively simple genotyping technologies for SNP detection rely largely on the polymerase chain reaction (PCR). In this procedure, two chemically synthesized single-stranded DNA fragments, or primers, are designed to match chromosomal DNA sequences flanking the segment in which an SNP is positioned. With the addition of nucleotide building blocks and a heat-stable DNA polymerase, the primer pairs, or amplicons, initiate synthesis of new DNA strands using the chromosomal material as a template. Each successive copying cycle, initiated by “melting” the resulting double-stranded products with heat, doubles the number of DNA segments in the reaction (Fig. 1-4). The technique is exceptionally sensitive; millions of identical DNA copies can be generated in a matter of hours with PCR using a single DNA molecule as the starting material. Even when the SNPs within a given haplotype are not directly involved in a disease, they provide markers for clonality and for the loss or rearrangement of specific chromosomal segments in growing tumors. In the human nucleus, each of the 23 tightly compacted chromosomes has a characteristic size and structure and a distinctive base sequence that carries unique protein coding information. Other noncoding DNA sequences are used for directing the transcription of neighboring genes through complex regulatory circuits involving protein binding and modification of the DNA itself, or shifting of its chromosomal packaging. Although genomic instability generally is considered a consequence of tumor formation rather than the initial trigger of cancer, the loss, gain, or rearrangement of chromosomal segments through deletion or translocation is a common form of neoplastic mutation, as protein-coding segments from different genes are combined or regulatory sequences are brought into new proximity to genes they do not normally control, as is seen in persons with chronic myeloid leukemia. In persons with chronic myeloid leukemia, recombination events lead to the fusion of BCR and ABL genes (Philadelphia chromosome). This process results in constitutive activation of the fused gene, leading to loss of proliferative control in myeloid cells and, consequently, cancer. Gross changes in DNA arrangement can be detected by cytogenetic analysis of chromosomal features on metaphase spreads. Fluorescent in situ hybridization provides greater resolution by localizing specific chromosomal DNA sequences corresponding to fluorescently labeled probes (Fig. 1-5) and can be used to track specific alterations in chromosomal structure where known genes are involved. Certain regulatory DNA sequences common to many genes are positioned upstream of the transcription start site (Fig. 1-6). Collectively called the “promoter” of a gene, these proximal sequences constitute binding sites for the RNA polymerase and its numerous cofactors. Whereas the position of the promoter with regard to the transcription start site is relatively inflexible, other DNA regulatory elements, known as enhancers, occur in unpredictable locations, often at a considerable distance from the genes they control. Some transcription factors bind to particular regions of enhancers and drive their associated genes in many types of cells, whereas others, which are active in only a limited variety of cells, maintain a tissue-specific pattern of gene expression. Enhancers often are responsible for the aberrant expression of genes induced by chromosomal translocation-associated specific forms of cancer; for example, a normally quiescent gene promoting cell growth that is dislocated to a position near a strong enhancer may be activated inappropriately, resulting in loss of control of growth.
Molecular Tools in Cancer Research
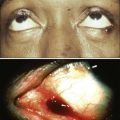
Detecting Cancer Mutations
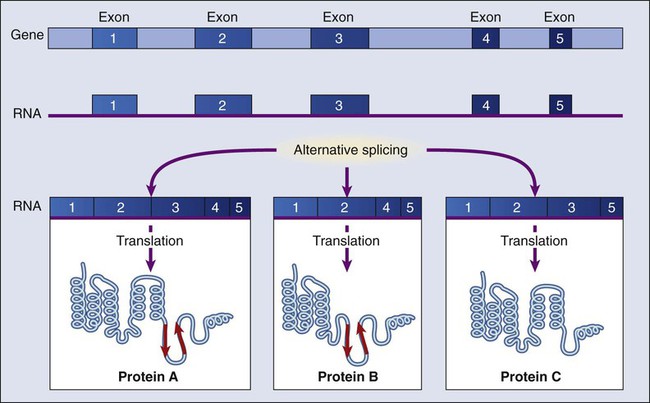
Generating Diversity with Alternate Splicing
The Genomics of Cancer
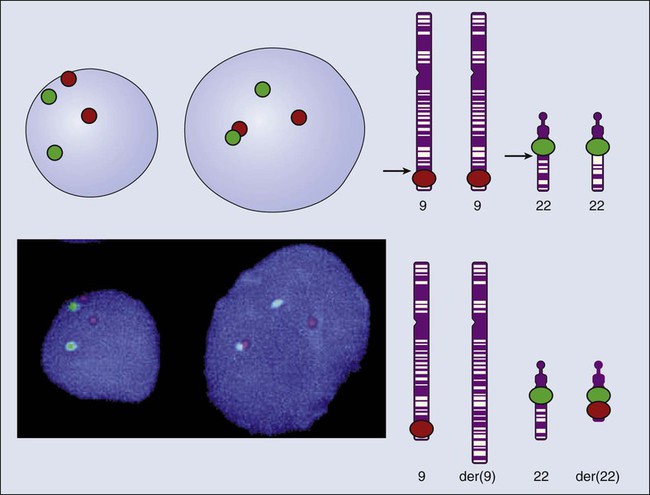
Losing Control of the Genome
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Oncohema Key
Fastest Oncology & Hematology Insight Engine