|
 Tyrosine receptor kinases and angiogenesis
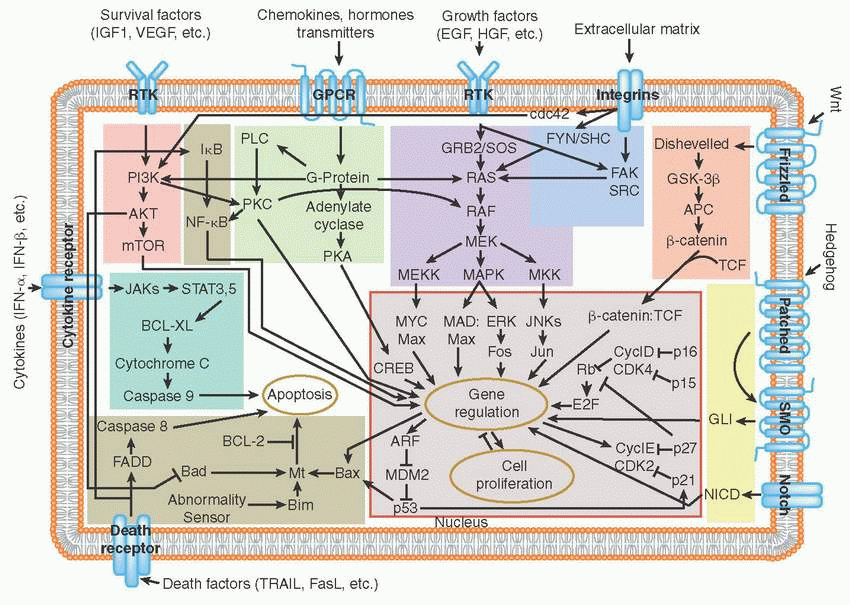
Tyrosine receptor kinases and angiogenesis G-protein-coupled receptor pathway
G-protein-coupled receptor pathway RAS/MAPK pathway
RAS/MAPK pathway PI3K pathway
PI3K pathway JAK/STAT pathway
JAK/STAT pathway Hedgehog and Notch pathways
Hedgehog and Notch pathways Wnt pathway
Wnt pathway SRC pathway
SRC pathway NF-χB pathway, death receptors and apoptosis
NF-χB pathway, death receptors and apoptosis Cell cycle regulation: TP53, RB1, aurora kinases
Cell cycle regulation: TP53, RB1, aurora kinases Activators (positive regulation)
Activators (positive regulation) Neither activating nor suppressing
Neither activating nor suppressing Suppressors (negative regulation)
Suppressors (negative regulation) Ligands
Ligands Cell membrane
Cell membrane Receptors
Receptors
|
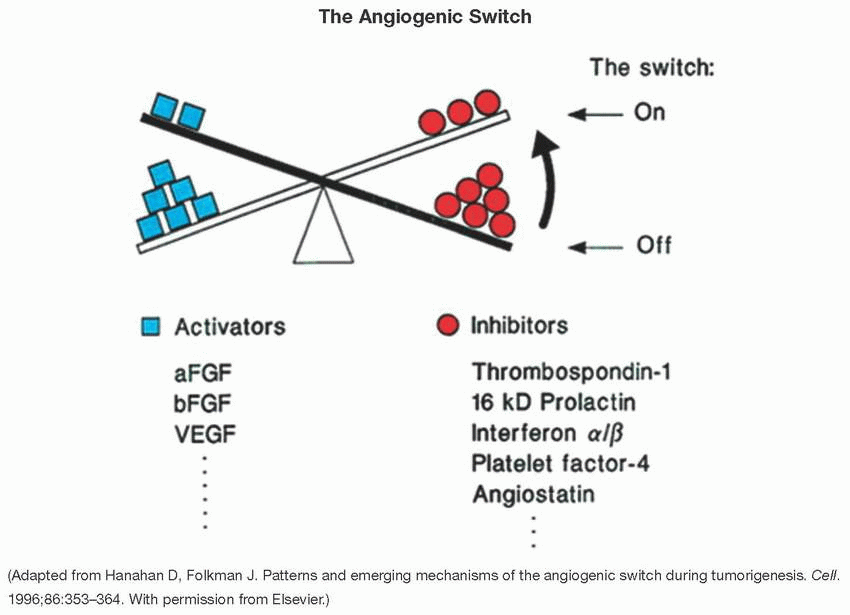
Angiogenesis is a hallmark of cancer represented by the development of new blood vessels to keep up with the tumor needs, a process that is tightly controlled by pro- and antiangiogenic chemical signals.
The angiogenic switch occurs when, in the hypoxic and inflammatory context of cancer, proangiogenic signals outweigh antiangiogenic signals, leading to tumor neovascularization. The shift in balance toward proangiogenic signals favors the abnormal and rapid growth and proliferation of local blood vessels.
The sprouting of new blood vessels from the existing vasculature is the most widely investigated mode of new vessel formation in tumors. There are five other mechanisms of new vessel recruitment:
Vasculogenesis involving vessel formation by endothelial progenitor cells (EPCs), which are recruited from the bone marrow and/or are resident in vascular walls.
Intussusception is the splitting of preexisting vessels to give rise to daughter vessels.
Vessel co-option occurs when cancer cells grow around and co-opt the existing vasculature.
Vascular mimicry is a process in which cancer cells get incorporated into the blood vessel wall.
Tumor stem cell to endothelial cells (ECs) differentiation occurs when cancer stem-like cells differentiate into ECs.
Tumor angiogenesis, which allows for continued tumor growth, is often a deregulated process that results in incomplete, irregular, tortuous, and leaky capillaries. Tumor vessels exhibit bidirectional blood flow, are not constantly perfused, and tend to be larger than normal vessels with an altered surface area-to-volume ratio that results in poor nutrient delivery and waste removal.
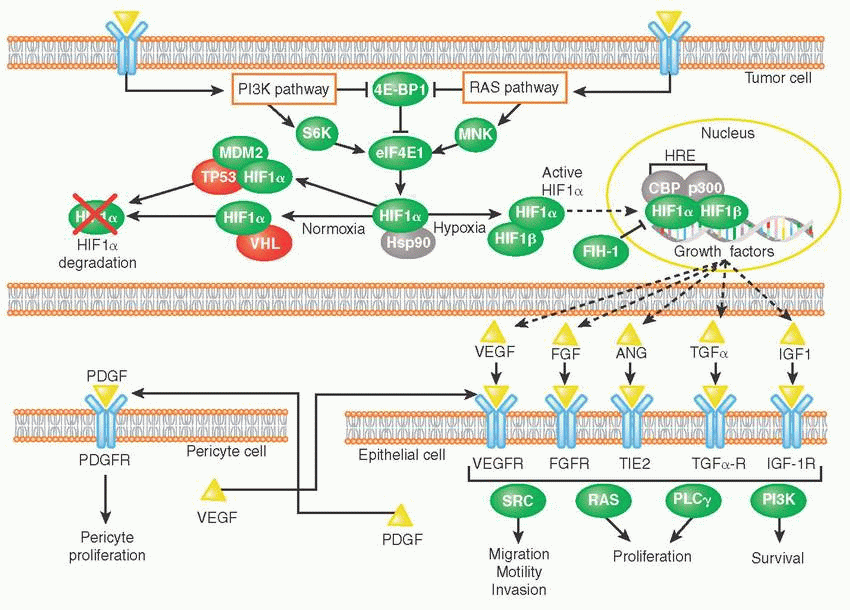
Hypoxia or low oxygen tension results from uncontrolled proliferation of cancer cells in the absence of a functional and adequate vascular bed. It is a major driver of tumor angiogenesis.
Under normoxic conditions, hypoxia-inducible transcription factor-1 alpha (HIF-1α) subunits are subjected to von Hippel-Lindau (VHL)-directed protein degradation. VHL itself is a tumor suppressor, and VHL mutations have been implicated in malignancies such as renal cell carcinoma.
Hypoxic conditions stabilize HIF-1α, allowing it to translocate to the nucleus, and, along with HIF-1β, initiate the transcription of proangiogenic genes such as VEGF. Examples of proangiogenic signals secreted by tumors include the following:
Growth factors such as fibroblast growth factor (FGF), angiopoietin (ANG), platelet-derived growth factor (PDGF), vascular endothelial growth factor (VEGF)
Cytokines such as interleukin-8 (IL-8)
TP53 negatively regulates angiogenesis by downregulating VEGF and other proangiogenic factors while increasing the expression of anti-angiogenic signals such as thrombospondin 1 (TSP1). Loss of P53 function promotes VEGF expression and tumor angiogenesis, although the underlying mechanisms remain unclear and controversial.
Other examples of endogenous inhibitors of angiogenesis include angiostatin, endostatin, tumstatin, and camstatin.
|
Induction of Tumor Angiogenesis | |||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||
| |||||||||||||||||||||||||||||||||||||
|
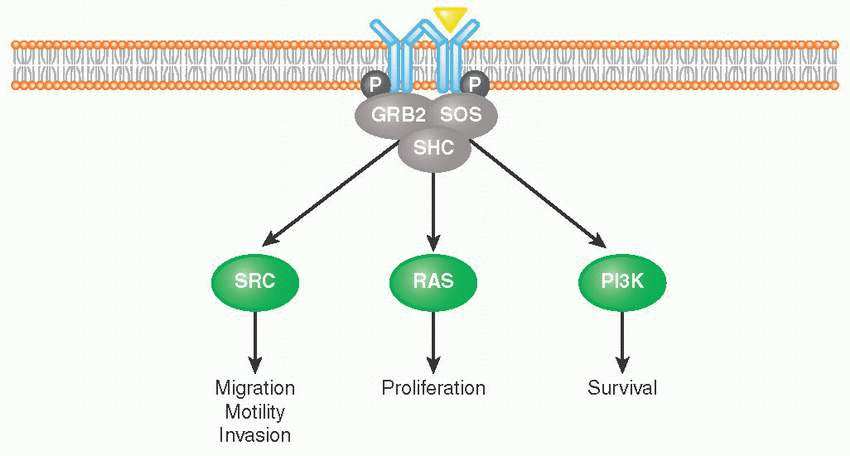
Receptor tyrosine kinases (RTKs) are cell surface receptors that function as key regulators of fundamental cellular processes such as proliferation, differentiation, senescence, and migration, among others.
Binding of growth factor ligands to extracellular domain, RTKs induces receptor dimerization (forming either homodimers or heterodimers with other RTKs) followed by their autophosphorylation. This allows for subsequent recruitment of adaptor proteins such as growth factor receptor-bound protein 2 (GRB2), SRC homology 2 domain-containing protein (SHC), and Son of Sevenless (SOS) that activate proliferation, survival, and migration pathways (e.g., RAS, PI3K, SRC).
Tight control of RTK activation is necessary for tissue homeostasis, which is why RTKs constitute one of the biggest classes of oncoproteins.
Over 20 different classes of RTKs have been identified, including the following that have been previously implicated in cancer initiation and progression: ALK, EGFR, FGFR, IGFR, KIT, MET, PDGFR, RET, TIE, and VEGFR. Each RTK will be discussed in detail in this section.
Vascular endothelial growth factor (VEGF) is one of the most important and prominent proangiogenic factors. VEGF’s normal function is to create new blood vessels during embryonic development, new blood vessels after injury, muscle following exercise, and new vessels (collateral circulation) to bypass blocked vessels.
The VEGF family consists of five members (VEGF-A, -B, -C, -D, and placental growth factor [PIGF]), which transmit signals via three VEGF receptors (VEGFR-1 through -3).
Many human cancers are found to overexpress VEGFs and/or VEGFRs.
VEGFR-1 binding by VEGF-B plays a role in the maintenance of newly formed blood vessels.
VEGFR-2 activation by VEGF-A binding has been shown to stimulate EC mitogenesis and cell migration, leading to cancer progression and metastasis.
VEGFR-3 is predominantly expressed in lymphatic vessels. When bound by either VEGF-C and VEGF-D, VEGFR-3 plays a role in lymphangiogenesis and metastatic spread to lymph nodes in a pathologic setting.
|
Fibroblast growth factor receptors (FGFRs) family has four highly conserved transmembrane RTKs (FGFR1-4) that differ in their ligand affinities and tissue distribution.
Fibroblast growth factors (FGFs) include 18 structurally related polypeptides (FGF1-10, FGF16-23) that signal through FGFRs.
FGFRs can bind canonical FGFs that are in complex with heparan sulfate proteoglycans (HSPGs) in an autocrine and paracrine fashion.
FGFRs can also bind endocrine FGFs (FGF19, 21, and 23) that are found in circulation, free of HSPGs.
Once bound to their ligands, FGFRs are induced to dimerize and cross-phosphorylate the tyrosine kinase domain on the cognate receptor.
Various downstream effector molecules are then recruited, allowing for the activation of signaling events that culminate in regulation of various cellular processes such as cell survival and proliferation, organ development, angiogenesis, and tissue homeostasis, to name a few.
Each FGFR can be activated by several FGFs; conversely, FGFs can activate more than one receptor.
Dysregulation of FGFR signaling in cancer may be caused by the following:
FGFR gene amplifications, activating mutations, translocations, and fusions
Amplification of FGF and FGF-related genes
Dysregulated FGFR signaling may contribute to cancer by the following:
Stimulating cancer cell proliferation
Driving tumor neovascularization
Promoting resistance to anticancer therapies
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
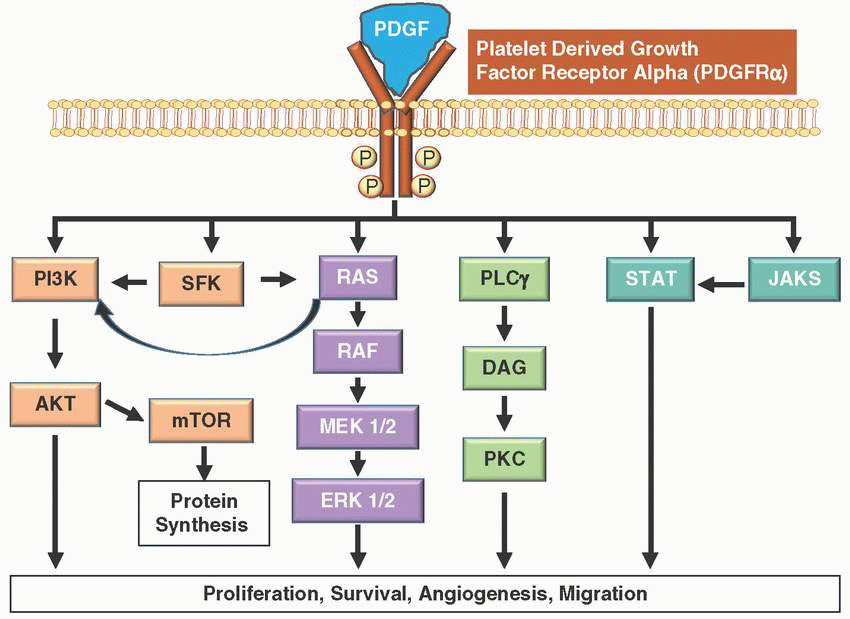
Platelet-derived growth factors (PDGFs) are potent mitogens for cells of mesenchymal origin and are synthesized, stored, and released by platelets upon activation; also produced by smooth muscle cells, activated macrophages, and ECs.
PDGF signaling network consists of four ligands (PDGF A-D) and two transmembrane tyrosine RTKs (PDGFRα and PDGFRβ).
These receptors transmit extracellular growth factor signaling to intracellular signaling cascades. Upon binding of the growth factor ligands, the receptors undergo dimerization and autophosphorylation of cytoplasmic kinase domains, activating the receptor.
Downstream effects of PDGFR activation leads to differential signaling through MAPK, PI3K, and/or JAK/STAT pathways resulting in cell proliferation, cell differentiation, survival, and migration.
Dermatofibrosarcoma protuberans is characterized by a chromosomal translocation with formation of COL1A1-PDGFB fusion gene, causing activation of the platelet-derived growth factor receptor beta (PDGFRB) in tumor cells.
PDGFRα is encoded by the PDGFRA gene, which is altered in multiple tumor types, resulting in constitutive PDGFRα activity.
Germ-line mutations in PDGFRA have been described in hereditary gastrointestinal stromal tumors (GIST).
PDGFRA amplification has been described in sarcomas and gliomas; however, often it is associated with co-alterations in EGFR, KIT, and KDR.
In about 5% of GIST, somatic PDGFRA mutations in exons 12, 14, and 18 activation loop are the most common and are associated with greater response to PDGFRA-inhibitor imatinib. However, up to 60% of PDGFRA mutant GIST harbors a mutation in exon 18 D842V, which is associated with primary resistance.
There are several other US Food and Drug Administration (FDA)-approved nonspecific multitarget tyrosine kinase inhibitors (TKIs) with multiple targets including PDGFRα and PDGFRβ such as axitinib, dasatinib, imatinib, lenvatinib, midostaurin, nilotinib, nintedanib, pazopanib, regorafenib, sorafenib, and sunitinib.
FDA-approved drugs targeting only PDGFRα and not PDGFRβ are ibrutinib, olaratumab, and ponatinib.
Imatinib is the first-line treatment option for GIST with PDGFRA mutations, followed by sunitinib for imatinib-resistant or -intolerant patients.
Second-generation PDGFRα inhibitors are in development to overcome resistance conferred by specific point mutations (e.g., p.D842V) such as DCC-2618 and BLU-285.
DCC-2618 is a potent pan-KIT and PDGFRα switch control inhibitor that has activity across a broad range of mutations that emerge on imatinib treatment, showing encouraging disease control in heavily pretreated GIST patients demonstrating objective responses in a phase 1 clinical trial.
Other small-molecule inhibitors targeting also PDGFRα and PDGFRβ currently in phase 3 clinical trials are anlotinib, cediranib, crenolanib, famitinib, masitinib, quizartinib, tivozanib, and valatinib.
|
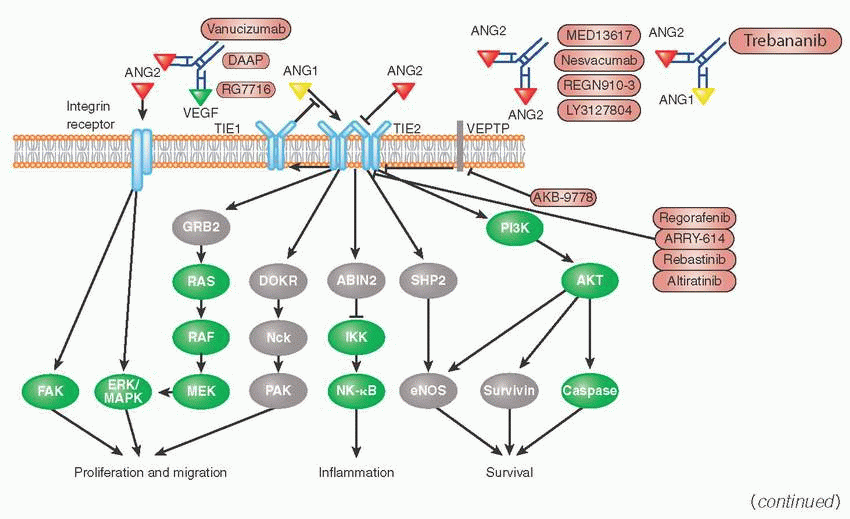
Angiopoietin growth factors (ANG) bind to cell surface RTKs with immunoglobulin-like and epithelial growth factor-like domains 1 and 2 (TIE1 and TIE2).
Four different ANGs have been identified (ANG1, ANG2, ANG3, and ANG4).
ANG1 and ANG4 are agonists for TIE2, whereas ANG2 and ANG3 are competitive antagonists for TIE1 and TIE2.
Activation of TIE2 by ANG1 is indispensable for embryonic cardiac development and angiogenesis, and both TIE1 and TIE2 are key regulators of normal formation of blood and lymphatic vessels development as TIE1 and TIE2 are almost exclusively expressed in the endothelium.
ANG/TIE system is also involved in pathologic processes including inflammation, metastasis, tumor angiogenesis, atherosclerosis, and vascular leakage.
Upon binding of ANG ligands, TIE2 receptors can form dimers or possibly multimers. Vascular endothelial protein tyrosine phosphatase (VEPTP, encoded by PTPRB gene) dephosphorylates active TIE2.
TIE1 upregulates the cell adhesion molecules (CAMs) VCAM-1, E-selectin, and ICAM-1 through a p38-dependent mechanism.
Regorafenib is an FDA-approved drug that inhibits TIE2.
ANG2-targeted investigational drugs include the peptide-Fc fusion protein trebananib (also known as AMG386), which blocks the binding of both ANG1 and ANG2 to TIE2. Trebananib was the first TIE inhibitor tested in a phase 3 clinical trial and showed an increased progression-free survival (PFS) in combination with paclitaxel in ovarian cancer, however, did not prolong the overall survival significantly.
Other monoclonal antibodies targeting ANG, such as MEDI3617, LY3127804, and nesvacumab (also known as REGN910), that block the interaction of ANG2 with TIE2 are currently in development.
Small-molecule TIE2 inhibitors under investigation include rebastinib and altiratinib.
|
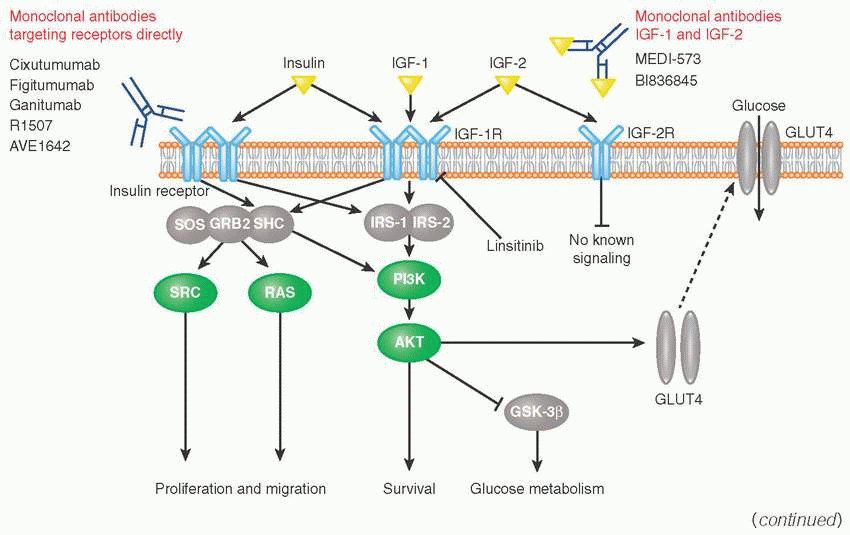
Insulin-like growth factor family includes two growth factors (IGF-1, IGF-2) and two receptors (IGF-1R, IGF-2R).
IGF-1 and IGF-2 are polypeptide hormones structurally similar to insulin. IGF-IR, IGF-2R, and insulin receptors are also structurally similar and are able to form heterodimers.
IGF-1R is activated by IGF-1, IGF-2, and insulin and has a strong growth-promoting effect.
IGF-2R does not trigger signaling, but regulates extracellular IRS-2 levels through receptor-mediated endocytosis and degradation. Upon phosphorylation, IGF-1R recruits and phosphorylates adaptor proteins IRS-1 (insulin receptor substrate), IRS-2, and SHC.
IRS-1 acts as a second messenger within cell to stimulate transcription of insulin-related genes (e.g., ELK1, GSK3) via PI3K and RAS pathways and also initiates the translocation of glucose transporter (GLUT) to the cell surface where it facilitates transport of glucose into the cell.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree