Genetic mutation
Mutation type
Melanoma subtypes affected
Melanocytic nevus subtypes affected
Type of test
KIT
Point mutation, amplification
Acral (~15 %), Mucosal (~15 %), CSD skin (~10–15 %)
Uncommon/rare
IHC
SS
NGS
FISH
CGH
BRAF
Point mutation, kinase fusion
Non-CSD skin (~60–70 %), CSD skin (~10 %), acral (~15 %)
Common acquired nevi (~86 %), Spitz nevi (<10 %), dysplastic nevi (~50–60 %)
IHC
SS
NGS
NRAS
Point mutation
CSD skin (~15–20 %), Non-CSD skin (~15 %), acral (~10–15 %), mucosal (~15 %)
Congenital nevi (~80 %)
SS
NGS
HRAS
Point mutation
Rare
Spitz nevi (~15–20 %), deep penetrating nevi (~6 %)
SS
NGS
GNAQ or GNA11
Point mutation
Uveal (90 %), blue nevus-like (~50 %)
Blue nevi (~87 %), nevus of Ota (6 %)
SS
NGS
CDKN2A
Point mutation or deletion
Non-CSD skin (<10 %)
Atypical Spitz tumors (?%)
SS
NGS
FISH
CGH
PTEN
Point mutation or deletion
Non-CSD skin (~10–30 %), CSD skin (<10 %)
Dysplastic nevi (?%)
IHC
SS
NGS
FISH
CGH
BAP1
Point mutation or deletion
Uveal (~47 %), cutaneous melanoma subtypes (?%)
“BAPoma” subset of Spitz nevi/tumors (100 %)
IHC
SS
NGS
Pathways Involved in Melanoma Pathogenesis
There are at least three major pathways associated with melanoma pathogenesis, namely:
1.
Mitogen activated protein kinase (MAPK)
2.
PI3K/AKT/MTOR pathway
3.
Retinoblastoma 1 (RB1) pathway
In addition to these pathways, other genes involved in melanocytic neoplasia include BRCA1 associated protein-1 (BAP1) in uveal melanomas and some cutaneous melanomas (and the BAP1 deficient tumor syndrome predisposing patients to uveal and cutaneous melanomas, mesotheliomas, and renal carcinoma, among others). More recently, kinase fusions involving anaplastic lymphoma receptor tyrosine kinase (ALK), BRAF, ret proto-oncogene (RET), and ROS proto-oncogene 1, receptor tyrosine kinase (ROS1) have also been identified as early initiating mutational events in Spitz tumors.
BRAF and the MAP Kinase Pathway
A key molecular pathway implicated in melanoma pathogenesis comprises RAS, RAF, MEK1/2, and ERK1/2, collectively referred to as the mitogen activated protein kinase (MAPK) which is involved in cell proliferation, survival, angiogenesis and invasion [4] (Fig. 17.1). Early mutations in the pathogenesis of melanocytic tumors are frequently in the mitogen activated protein kinase (MAPK) pathway including in particular B-Raf proto-oncogene (BRAF), neuroblastoma RAS viral (v-ras) oncogene homolog (NRAS), Harvey rate sarcoma viral oncogene homolog (HRAS), Guanine nucleotide binding protein (G protein) and q polypeptide (GNAQ) and guanine nucleotide binding protein (G protein), alpha 11 (Gq class) (GNA11), collectively known as GNAQ/GNA11, and v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT) (Fig. 17.1). These mutations are typically mutually exclusive of each other and are found in particular subtypes of melanocytic nevi and melanomas. The binding of ligand or factor to the cell surface receptor causes phosphorylation and activation of son of sevenless 1 (SOS1), which promotes removal of GDP from NRAS, allowing it to bind GTP in its active form. Activated NRAS promotes the protein kinase BRAF (see below) which in turn phosphorylates MEK (MEK1 and MEK2) with downstream phosphorylation and activation of a mitogen activated protein kinase (MAPK), also known as extracellular signal regulated kinase (ERK) [5, 6].
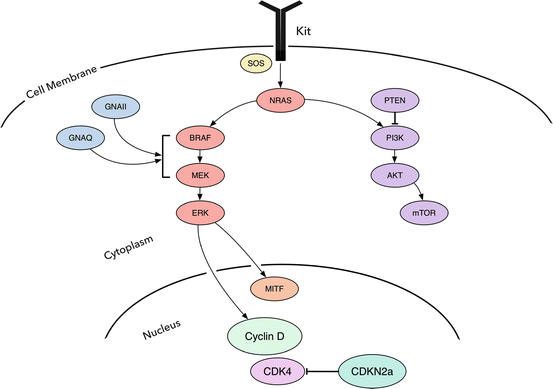
Fig. 17.1
The major signaling pathways in melanoma are the MAPK signaling pathway and the AKT/PI3K pathway, which are involved in cell proliferation, growth, and cell death
A commonly tested member of this pathway is BRAF, a proto-oncogene on chromosome 7 which encodes for the BRAF protein. BRAF protein has three domains, a Ras–GTP-binding self-regulatory domain, a serine-rich hinge region, and a catalytic serine/threonine kinase domain that transmits signals from RAS to MEK1/2 [4, 7, 8]. Mutational activation of BRAF in melanocytes is important in the initiation of neoplasia, due to its involvement in cellular proliferation, invasion and angiogenesis but it is insufficient for progression to melanoma. Since BRAF directly phosphorylates another kinase MEK, MEK inhibitors are of utility in BRAF mutated tumors resistant to BRAF inhibitor therapy (see below). Approximately 90–97 % of mutations are a substitution of thymine to adenine at nucleotide 1796 resulting in a substitution of valine (V) to glutamate (E) at codon 600 (V600E). Other mutations in exon 15 include V600K, V600R, and exon 11 can show G469V and G469R point mutations [4, 9, 10]. Mutations in the kinase domain can result in the production of a protein that is ten times more active than wild type BRAF. BRAF mutations can be found 50–70 % of melanoma as well as 86 % of melanocytic nevi, and less so in Spitz nevi [4, 11–13] (Table 17.1).
The V600E mutation in BRAF can be detected by immunohistochemistry (IHC). In a recent study the detection of BRAF V600E mutation via IHC with confirmation using direct DNA sequencing or allele specific PCR demonstrated a sensitivity of 100 % and specificity of 97 % [14]. Other studies show similar findings [15, 16]. The IHC antibody is highly specific for the V600E mutation and will not detect the presence of a V600K or V600R mutation. Although IHC for BRAF is seen as a promising surrogate for detection of the V600E mutation, currently the only FDA approved platforms for the detection of the V600E mutation rely on targeted sequencing analysis or allele specific polymerase chain reaction (PCR) (a list of current FDA approved molecular diagnostic tests can be found through the FDA website and is also available on the Association for Molecular Pathology website), thus limiting the use of this antibody in daily practice. Next-generation sequencing is also utilized for BRAF mutational analysis (typically for panel testing in association with assessment for multiple genetic mutations).
The treatment of melanoma with a BRAF mutation includes BRAF inhibitors such as vemurafenib, approved in 2011 for patients with BRAF V600E positive stage III or IV metastatic melanoma [17]. Treatment with vemurafenib is discouraged in wild type BRAF or more upstream NRAS mutant metastatic melanoma, due to paradoxical activation and tumor promotion via the MAPK pathway [18, 19] highlighting the importance of mutational testing (for NRAS as well as BRAF) prior to treatment. Other treatment possibilities include selective kinase inhibitors such as dabrafenib which was approved in 2013 [20]. Downstream MEK1/2 inhibitors such as trametinib and cobimetinib have shown improved survival with BRAF mutated tumors or melanomas with resistance to BRAF inhibitors. Although monotherapy for MEK inhibitors has not been approved, the FDA has approved combination dabrafenib–trametinib for patients with a BRAFV600 mutation [21–24].
NRAS
The NRAS (a member of RAS GTPase family) gene is located on chromosome 1 with downstream effects on MEK and AKT with release of nuclear transcription factors causing cellular proliferation and apoptosis [25] (Fig. 17.1). NRAS mutations are found in approximately 15 % of melanomas from intermittently sun-exposed skin, approximately 15–20 % of melanomas from chronically sun-damaged skin, about 10 % of acral melanomas, and in approximately 15 % of mucosal melanomas [26]. NRAS mutations can also be seen in 64–81 % of congenital nevi [27]. Mutations in codon 16 comprise approximately 90 % of alterations seen in melanoma [28, 29]. Typically melanomas harbor either a BRAF or NRAS mutation, although the two are not completely mutually exclusive [4, 30, 31]. Unlike BRAF mutations that require a synchronous mutation in the PI3K/mTOR pathway, NRAS mutations are able to simultaneously activate both the MAPK and PI3K/MTOR pathways. Mutations in both BRAF and NRAS genes are more commonly seen in melanomas found in areas with intermittent exposure, such as trunk and extremities and less so in areas of chronic sun exposure such as the head and neck [28]. No NRAS targeted treatments are available; however, downstream MEK inhibitors such as MEK162 have shown improvement in tumor progression in recent clinical trials [32]. NRAS mutational analysis in conjunction with BRAF mutational analysis is often performed to exclude the possibility of paradoxical tumor progression secondary to BRAF-inhibitor therapy in patients with tumors harboring both mutations simultaneously.
HRAS
HRAS, another RAS GTPase family member is involved in cell progression and growth [33]. Copy number gains and/or mutations in HRAS are rare in melanoma; however, they are found in up to 20 % of Spitz nevi (Table 17.1) [34, 35]. Mutations are typically found in either exon 2 (codon 12 or 13) or exon 3 (codon 61). Spitzoid tumors harboring HRAS mutations tend to be mainly intradermal with an infiltrative growth pattern and desmoplasia and preliminary data suggest that they behave in a benign fashion [35, 36]. Interestingly, HRAS mutations have also been shown in deep penetrating nevi suggesting that these unusual nevi may be variants of Spitz nevi [37]. Amplification of the short arm of chromosome 11 (11p) can be detected via comparative genomic hybridization (CGH) or fluorescence in situ hybridization (FISH) and HRAS mutation via targeted sequencing analysis. Although due to the rarity of HRAS mutation in melanoma, detection of an HRAS mutation in a Spitzoid lesion may serve to confirm the diagnosis of Spitz nevi over melanoma, in practice this type of analysis does not play a routine role.
KIT
The KIT (v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog) gene is located on the long arm of chromosome 4 and the product is a type III receptor tyrosine kinase (RTK) CD117 or cKIT. When activated, dimerization and activation of its intrinsic tyrosine kinase activity leads to downstream phosphorylation of both the MAPK pathway and the AKT/mTOR pathway (Fig. 17.1). KIT amplification or mutation can cause constitutive activation and subsequent cellular proliferation.
KIT mutations are found in approximately 15 % acral melanomas, 15 % of mucosal melanomas and 10–15 % of melanomas found on chronically sun-damaged skin [26, 38] (Table 17.1). Most mutations are found in exon 11 with a L576P substitution. Other mutations can be seen in exons 13 and 17 [39–41]. KIT amplifications are found in 25–30 % of acral and mucosal melanomas [42]. Discordance in the immunohistochemical detection of the KIT protein with available anti-CD117 (rabbit polyclonal) antibodies, as compared to mutational analysis by gene sequencing of exon 11, 13, and 17 is well known, making IHC a poor surrogate for detection of the mutation or amplification by PCR [40, 41].
GNAQ/GNA11
Uveal melanomas have a near complete absence of BRAF and NRAS mutations and rather show mutations in GNAQ and GNA11. GNAQ and GNA11 genes encode for the alpha subunit of a heterotrimeric G-protein, located on chromosomes 9 and 19 in humans, respectively. G proteins are involved in transducing extracellular signals via transmembrane G-protein coupled receptors which cause conformational changes in G-proteins mediating signals to downstream effectors such as phospholipase C (PLC) [38, 45, 46] (Fig. 17.1). Up to 90 % of uveal melanomas contain GNAQ or GNA11 mutations, usually in codon 209, which is essential for GTP hydrolysis. Over 80 % of blue nevi and 6 % nevus of Ota also contain mutations in GNAQ or GNA11 (Table 17.1). Interestingly, the presence of a nevus of Ota is a known risk factor for uveal melanoma and harbors the same mutation in a subset of cases. Melanoma arising in a blue nevus (so-called “malignant blue nevus”) also has been shown to harbor GNAQ or GNA11 mutations. GNAQ is similar to BRAF and NRAS in that it is a mutation expressed in nevi as well as melanomas and is insufficient alone for progression to melanoma. Overall, approximately 90 % of all uveal melanomas harbor mutations in either GNAQ or GNA11 genes, with 95 % of the mutations occurring at Q209 and 5 % occurring at R183 [3]. Since GNAQ and GNA11 mutations are involved in the MAPK pathway, in vitro studies examining the utilization of a combination of a downstream MEK inhibitor and mTOR inhibitor [47, 48] show some promise [45, 49].
PI3K/AKT/MTOR Pathway
A second pathway that runs parallel to the MAPK pathway involves phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha (PIK3CA), v-akt murine thymoma viral oncogene homolog 1 (AKT1), mechanistic target of rapamycin (MTOR) collectively known as the PI3K/AKT/MTOR pathway (Fig. 17.1). Phosphatase and tensin homolog (PTEN) is involved at the end of this pathway. Mutations or variants in this pathway tend to occur later in tumor progression.
PIK3CA is a lipid kinase activated by downstream effects of an active growth factor receptor tyrosine kinase (Fig. 17.1). AKT1, a serine/threonine kinase interacts with the phospholipids produced by PIK3CA and translocates to the inner membrane. Activated AKT1 modulates the function of numerous substrates involved in the regulation of cellular growth and proliferation including MTOR, B-cell CLL/lymphoma 2 (BCL2), NFKB activating protein (NKAP), cyclin-dependent kinase inhibitor 1B (CDKN1B also known as p27), and MDM2 proto-oncogene E3 ubiquitin protein ligase (MDM2). MTOR, a threonine-protein kinase is located on chromosome 1 and activates more downstream molecules involved in cellular proliferation. PTEN located on chromosome 10 acts as a tumor suppressor encoding for a lipid phosphatase that negatively regulates the PI3K/AKT/mTOR pathway by promoting apoptosis [50].
PTEN mutations can be found in up to 30 % of melanomas [51]. PTEN inactivation can be detected via IHC or targeted sequencing. As there are currently no FDA approved therapies for treatment of melanomas with PTEN mutation and PTEN analysis is not yet routine in clinical practice, however, PTEN loss may play a role in resistance to BRAF inhibitors in BRAF mutated melanomas; however, testing may become routine in the future [52].
Retinoblastoma (RB1) Pathway
A third important pathway plays a key role in cell cycle regulation, which is the retinoblastoma 1 (RB1) pathway. Key genes involved in melanoma pathogenesis in this pathway include tumor protein p53 (TP53), cyclin-dependent kinase inhibitor 2A (CDKN2A), and cyclin-dependent kinase 4 (CDK4) (Fig. 17.1). Notably, germline mutation in either CDKN2A or CDK4 results in the hereditary dysplastic nevus melanoma syndrome.
CDKN2A (also known as p16) is located on chromosome 9p and is the most common mutated gene (40 %) in hereditary melanoma and the dysplastic nevus syndrome [60]. Importantly, somatic mutations in CDKN2A and/or loss of 9p21 are frequently seen in sporadic melanomas, the detection of which can play a key role in melanoma diagnosis. It encodes for p16, a tumor suppressor which binds to and inhibits CDK4, a kinase that phosphorylates retinoblastoma (RB1) protein and eventually leads to progression in the cell cycle from G1 to S phase. An altered p16 cannot inhibit CDK4, causing activation of the cell cycle. CDKN2A also encodes for p14, another tumor suppressor that inhibits MDM2 from degrading p53, which is involved with cell death and apoptosis. Mutated p14 promotes degradation of p53 preventing cell death and promoting cell survival [61]. CDK4 is located on chromosome 12 and is the catalytic subunit of a Serine/Threonine protein kinase that activates RB1 protein. It is mutated in 2–3 % of hereditary melanoma families [60]. Mutations in CDK4 have the same downstream effects of activation of the cell cycle as alterations in tumor suppressor p16 mentioned above [61]. Genetic testing via a blood sample or buccal mucosa swab for germline mutation in either CDKN2A or CDK4 via targeted sequencing analysis is currently available through several commercial laboratories. Testing is generally reserved for those individuals with a strong family history of melanoma.
BAP1
BRCA1-associated protein 1 (BAP1) is a tumor suppressor gene located on chromosome 3. The product is identified as an ubiquitin hydrolase causing ubiquitination of proteins involved in cellular growth [53, 54]. It is deleted in approximately 47 % of primary uveal melanomas and loss of BAP1 has been noted in melanocytic nevi with a nodular pattern or epithelioid cytology, so-called “BAPomas” [55–57] (Fig. 17.2). Recently, a tumor syndrome associated with germline mutation in BAP1 has been identified, predisposing patients to uveal and cutaneous melanoma, mesothelioma, renal cell carcinoma, and the previously mentioned epithelioid cell nevi/tumors [57, 58]. BAP1 mutational status in strongly associated with protein expression. BAP1 is ubiquitously expressed in the nucleus, with loss of nuclear expression seen in BAP1 deleted tumors. It has been suggested that immunohistochemical evaluation of uveal melanomas by BAP1 IHC should become routine [59]. Of note, BAP1 detection via IHC is also utilized to detect BAP1 deficient epithelioid cell (Spitz) tumors, which may arise in sporadic setting, but when multiple, may be a cutaneous marker for the presence of an underlying germline mutation in the gene. Testing for germline mutation in BAP1 is currently performed solely in the academic setting.
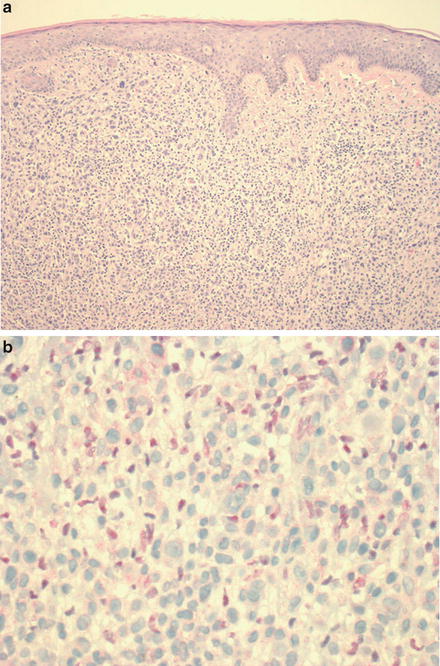
Fig. 17.2
(a) Typical BAP1 deficient epithelioid cell (spitzoid) melanocytic tumor from a patient with a germline mutation in BAP1 (H&E stain), with a patchy lymphocytic host response. (b) The large epithelioid melanocytes are negative for BAP1 by immunohistochemical staining (the scattered admixed smaller lymphocytes show positive nuclear staining for BAP1)
In summary, molecular testing in melanomas is fairly well established, mainly for treatment of advanced stage melanoma, including clinical trial enrollment. There are ongoing research and clinical trials evaluating other molecular biomarkers with potentially clinically utility. Evaluation for mutations via allele-specific PCR amplification and detection or targeted sequencing remain the most commonly utilized techniques, with panel testing via next-generation sequencing also becoming increasingly available both commercially and in the academic setting. Laboratories offering molecular diagnostic tests use either FDA-approved kits or an in-house test (so-called laboratory developed test, or LDT). While each assay type has its advantages and disadvantages (which are beyond the scope of this chapter), increasingly, determination of which assay type to use is being governed by governmental regulatory guidelines rather than assay specificity, as these regulations also affect insurance coverage and reimbursement for these tests.
Role of Molecular Biomarkers in Diagnosis
Most typical melanocytic lesions can be diagnosed as benign or malignant on light microscopy (Fig. 17.3). However, there are specific subsets of challenging melanocytic tumors/lesions with conflicting borderline histologic findings that preclude a definitive diagnosis based on morphologic grounds alone. These include:
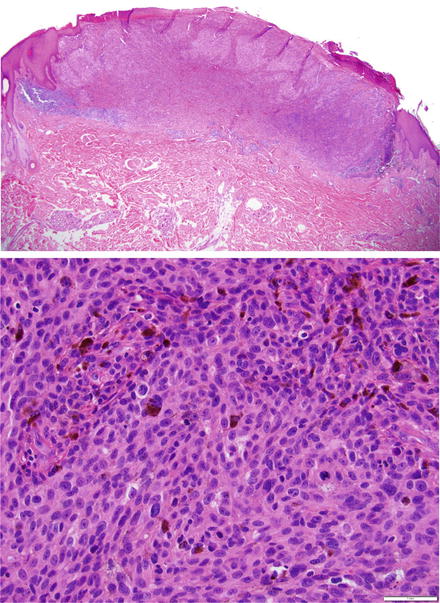
Fig. 17.3
(a) Invasive melanoma. Typical ulcerated invasive melanoma with (b) monotonous melanoma cells lacking maturation with numerous mitosis shown at higher magnification
Atypical spitzoid melanocytic proliferations,
Nevoid melanomas,
Spindle cell melanomas mimicking atypical fibroxanthomas or other fibrohistiocytic lesions,
Proliferative nodules versus melanoma in large congenital nevi,
Melanoma versus clear cell sarcoma,
Identification of residual melanoma in situ on severely sun damaged skin,
Melanosis versus regressed melanoma, and
Melanoma transformation within a dysplastic or other type of atypical nevus.
The lack of concordance based on morphologic assessment in melanocytic tumors with borderline histopathologic findings is notoriously high, frequently over 25 % [62], even among experts. This is particularly true for spitzoid lesions where at times diagnostic consensus cannot be reached and the clinical behavior of the tumor cannot be predicted with certainty [33, 63, 64] (Fig. 17.4). The morphologic limitations in the diagnosis of these histologically borderline lesions lead to both under and over diagnosis of melanoma. Ancillary immunohistochemistry or molecular testing may play a significant role in aiding diagnosis.
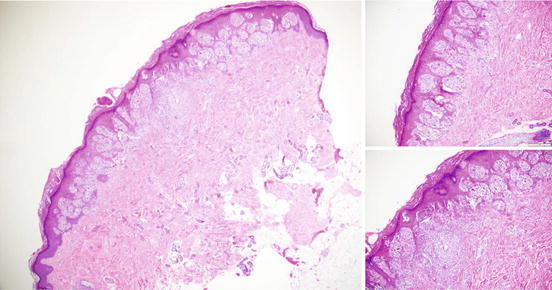
Fig. 17.4
Typical Spitz Nevus. A spitz nevus demonstrating symmetry with clefting of spitzoid nests (top right) and focal pagetoid upward migration (lower right) which can be seen in spitz
Immunohistochemistry
Currently, immunohistochemistry plays a first line role in the diagnostic work of melanocytic tumors, given its easy accessibility in most laboratories and relatively low cost. Evaluating expression of immunohistochemical markers highlighting melanocytic differentiation (Melan-A/MLANA, MITF, Mart-1, S100, SOX10) and proliferation indices (KI-67, PHH-3) is generally accepted. HMB45, a monoclonal antibody against gp100, shows loss of expression with depth/maturation in nevi in general and shows less of a gradient in melanomas. However, the application of HMB45 in particular is limited due to patchy expression around hair follicles, areas of lymphocytic inflammation, trauma, and prior biopsy. In addition, some nevic subtypes can show diffuse strong positivity for HMB45 (for example deep penetrating nevi, spitz nevi and blue nevi) similar to melanomas, and not all melanomas show HMB45 staining, or show staining similar to that seen in nevi. In addition, none of these markers is specific for melanocytic differentiation, as many of these markers can show expression in melanophages, mast cells, fibroblasts, dendritic cells as well as non-melanocytic lesions (neural, fat, cartilage), hampering their utility in more challenging lesions, such as intradermal atypical amelanotic tumors or metastases.
There have been attempts to identify biomarkers able to distinguish melanoma from nevi and or add prognostic value in melanoma staging [65]. However, no definitive single or panel of markers has been clearly identified and validated. This is not surprising given the genetic heterogeneity of melanomas in general as well as the frequent lack of direct correlation between protein expression at the IHC level and presence or absence of a given mutation or chromosomal aberration. Nonetheless, several antibodies in addition to the ones just mentioned may be used in select melanocytic tumor subtypes (Table 17.2). As mentioned in the previous section, BAP1 IHC is used to assess for BAP1 loss in a subset of atypical epithelioid cell (Spitz) tumors in order to classify them as “BAPomas”. BAPomas, while frequently showing epithelioid cytological atypia and occasional mitoses, appear to behave in an indolent fashion provided the only aberration is isolated BAP1 mutation and/or loss of chromosome 3p on which BAP1 resides [65]. These melanocytic nevi/tumors almost always also harbor a BRAF mutation, and thus BRAF IHC may also be utilized in the diagnostic evaluation of these lesions, as BRAF mutation is uncommon in other types of epithelioid cell (Spitz) tumors. Recently, kinase fusions have been identified in other Spitz tumor subtypes, including fusions with ALK1, ROS1, NTRK1, RET, BRAF, and NTRK3 [66]. An ALK antibody is commercially available and can be utilized as well in the diagnostic evaluation of Spitzoid lesions, although since ALK fusions have been identified in the full spectrum of Spitzoid lesions (i.e., nevi, atypical Spitz tumors, and spitzoid melanomas), it is currently of limited diagnostic utility by itself due to overexpression.
Table 17.2
Immunohistochemical markers used in the diagnosis of melanocytic lesions
Antibody | Antigen | Staining | Interpretation | Limitation |
|---|---|---|---|---|
SOX10 | SOX10 | Nuclear | Diffuse expression in melanoma and nevi, particularly helpful in desmoplastic melanoma | Sensitive but not specific (expressed in other tissues, including neural lesions) |
S100 | S100 (calcium binding protein) | Nuclear and cytoplasmic | Diffuse expression in both nevi and melanoma, especially helpful in desmoplastic melanoma | Sensitive but not specific (expressed in other cell types including fat, cartilage, dendritic cells, neural lesions) |
Melan-A; MART-1 | Melanoma antigen recognized by T cells 1 | Cytoplasmic | Diffuse expression in nevi, tends to be more patchily expressed melanoma | More specific but less sensitive (negative in up to 15 % of melanomas, particularly desmoplastic melanomas), can also be expressed in other tissues (i.e., melanophages, PEComas) |
HMB45 | Gp100 | Cytoplasmic | Gradient of expression in nevi (except blue nevi), more diffuse patchy expression in melanoma | Lower sensitivity than other melanocytic markers, also expressed in PEComas |
MiTF | Microphthalmia transcription factor (MiTF) | Nuclear | Diffuse expression in nevi and melanoma | Relatively low specificity (expressed in fibroblasts, Schwann cells, mast cells, PEComas, cellular neurothekeoma), usually negative in desmoplastic melanoma |
Tyrosinase | Tyrosinase | Cytoplasmic | Diffuse expression in nevi and melanoma, patchy expression in melanoma | Fairly specific for melanocytic differentiation, but less sensitive, also expressed in PEComas, negative in desmoplastic melanoma |
Ki67 (MIB1) | Ki-67 | Nuclear | proliferation marker, <1 % in nevi, generally >5 % in melanoma | Low sensitivity as stand-alone marker, also increased proliferation in nevi in children, pregnant women, proliferative melanocytic nodules within congenital nevi |
p16 | p16 | Nuclear and cytoplasmic | Complete absence of expression tends to correlate with clinically aggressive behavior in atypical Spitz tumors | There is a lack of direct correlation with p16 expression at the IHC level and biologically significant inactivation/deletion of CDKN2A |
ALK | ALK | Nuclear and cytoplasmic | Detects ALK rearrangements in Spitzoid neoplasms | An ALK rearrangement does not distinguish between indolent Spitz tumors and aggressive ones (Spitzoid melanomas) as they can be found in both |
BAP1 | BAP1 | Nuclear | Expressed in most nevi and majority of melanomas, loss of expression in BAPomas/subset of atypical Spitz tumors, and subset of uveal and cutaneous melanomas | Loss of BAP1 helps to subclassify atypical spitzoid lesions and has prognostic value in uveal melanoma; however, it does not distinguish between melanoma and atypical Spitz tumor by itself |
BRAF V600E | V600E (clone VE1) | Cytoplasmic | Sensitive and specific for the BRAF V600E mutation | Staining can be heterogeneous, the antibody will only detected V600E mutations in BRAF |
Finally, assessment for P16 expression via IHC may also be of use in the diagnostic workup of melanocytic tumors with borderline morphologic features, in particular atypical Spitz tumors, as homozygous deletion of 9p21 (where p16 resides) has been correlated with aggressive clinical behavior of atypical Spitzoid lesions [67–69]. However, due to heterogeneity in expression of p16 and clonal subpopulations, p16 expression does not always directly correlate with homozygous loss of 9p21.
Comparative Genomic Hybridization (CGH)
Melanomas in general (>95 % of primary melanomas) harbor frequent chromosomal aberrations in comparison to benign nevi [34, 70–73]. Differences in recurring chromosomal aberrations are seen in different melanoma subtypes (Table 17.3). Comparative genomic hybridization (CGH) compares tumor DNA to normal DNA, allowing for the detection of chromosomal alterations that lead to DNA copy number changes. CGH requires normal and abnormal tissue by the nature of its methodology and may require microdissection to accurately separate the two [74]. CGH is a molecular cytogenetic technique that analyzes copy number variation by comparing the chromosomal or subchromosomal (5–10 megabases) differences between the tumor and normal tissue by differentially labeling fluorophores and competitively cohybridizing onto to nucleic acid targets in metaphase. Array CGH improves resolution (5–10 kilobases) and allows for evaluation of the entire genome with a similar technique but with cohybridization onto DNA targets affixed to a solid platform [75]. Array CGH can be used to accurately quantify DNA copy number at the genomic level. It can be done on paraffin-embedded tissue and can detect single copy number deletions and duplications [76]. The advantage of aCGH is its ability to scan the whole genome for copy number changes. The use of CGH and more recently array CGH to detect chromosomal aberrations in ambiguous melanocytic tumors, however, is currently performed predominantly in the academic setting, due to the cost and relatively laborious nature of the assay. Another limitation of the assay is its relatively low resolution for identifying copy number changes, as those changes must be present in 30–50 % of the cell population in order for it to be detected [77].
Table 17.3
Common chromosomal aberrations in melanomas by anatomic location
Acral melanoma | Non-CSD (superficial spreading/nodular) | CSD (lentigo maligna melanoma) | |
|---|---|---|---|
Chromosomal gains | 1q, 6p, 7, 8q, 17q, 20q | 6p, 7q, 8q, 17q, 20q | 1q, 6p, 9q, 11q13, 15q, 17q, 20q |
Chromosome amplifications | 4q, 5p13, 5p15, 11q13, 12q14 | ||
Chromosomal losses | 6q, 9p, 9q, 10p, 10q, 11q, 15q, 16q, 21q | 9p, 10p, 10q, 21q | 6q, 8p, 9p, 13q, 17p, 21q |
Gene amplifications | CCND1, CDK4 | CCND1 | |
Tumor suppressor gene losses | CDK2NA, MDM2, PTEN | CDK2NA, PTEN | CDK2NA |
Fluorescence In Situ Hybridization (FISH)
The identification of recurring chromosomal gains, losses, and amplifications via CGH led to the development and validation of a set of FISH probes to aid in the diagnosis of melanoma in histologically ambiguous cases [68, 69, 78–80]. Fluorescence in situ hybridization is used to detect the presence and location of certain DNA sequences (genes) by complementary binding of a fluorescent probe to intact chromosomes in cells that are fixed to a slide, and has the advantage of lower cost, higher resolution, and ease of use. Based on available CGH and microarray data a set of four FISH probes that could accurately distinguish melanomas from nevi with a sensitivity of 86.7 % and a specificity of 95.4 % were initially developed (Table 17.4) and (Fig. 17.5). The following probes, RREB1 (6p25), centromere 6, MYB (6q23), and CCND1 (11q13) were found to be predictive of melanoma when: (1) more that 38 % of lesional nuclei contained >2 11q13 signals or, (2) >55 % of nuclei contained more 6p25 than centromere 6 signals, or (3) more than 40 % of nuclei demonstrated less 6p23 than centromere 6 signals, or (4) >29 % of nuclei contained >2 6p25 signals. This set of 4 four probes was also able to distinguish melanoma from adjacent nevic precursor in 78 % of cases (28/36 cases) [81]. These probes were also utilized to accurately distinguish, based on the algorithmic approach detailed above, nevoid melanomas from mitotically active nevi in 10/10 cases of nevoid melanoma, with 100 % sensitivity and 100 % specificity [78]. Using this same approach, epithelioid blue nevi were also able to be distinguished from blue nevus-like cutaneous melanoma metastases with high sensitivity (90 %) and specificity (100 %) [82].
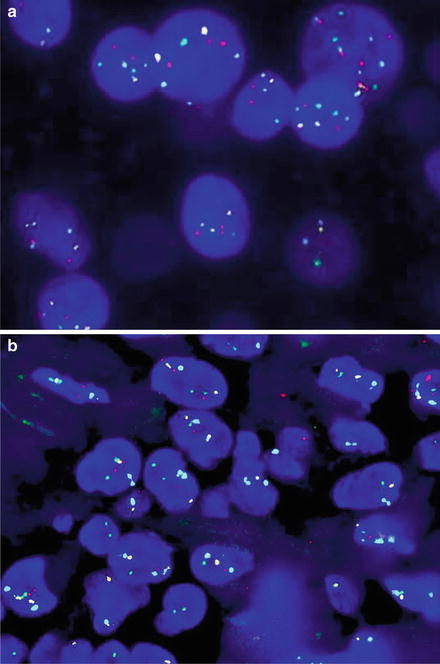
Table 17.4
FISH probes and their criteria for an abnormal signal and percentage cutoffsa
Probe | Gene | Function | Criteria | Abnormality | Cutoff |
|---|---|---|---|---|---|
6p25 | RREB1 | Zinc finger protein; ras responsive element binding protein 1 | >2 signals | Copy number gain | >29 % |
Centromere 6 | (CEP6) | Mitosis | RREB1 > CEP6 | Copy number gain | >55 % |
6q23 | MYB | Transcription factor; myeloblastosis viral oncogene homolog | MYB < CEP6 | Deletion | >40 % |
11q13 | CCND1 | Cyclin D1 (bcl-1) cell cycle regulator | >2 signals | Copy number gain | >38 % |
9p21 (with centromere 9 (CEP9)) | CDKN2A | Locus for p16 tumor suppressor | Homozygous deletion (using CEP9 as internal control) | Deletion | >33 % |
Fig. 17.5
(a) Melanoma with a positive melanoma fluorescence in situ hybridization (FISH) profile, with gains in RREB1 6p25 (red) and CCND1 11q13 (green). No loss of 6q23 Myb (yellow) or 9p21 (not shown) was seen. CEP 6 is the aqua hybridization signal. (b) A histologically borderline atypical melanocytic proliferation that was negative on the melanoma FISH assay (RREB1 6p25 (red), CCND1 11q13 (green), 6q23 Myb (yellow), CEP 6 (aqua))
More recently, homozygous deletion of 9p21 was found in unequivocal spitzoid melanomas and the presence of homozygous deletion of 9p21 in atypical Spitz tumors has also been correlated with aggressive behavior [67–69]. Homozygous deletion of 9p21 (using CEP9 as a control) of >33 % is considered significant in this assay according to the literature (although cutoffs for each probe may vary according to an individual laboratory validated melanoma FISH assay). The sensitivity and specificity of this six probe assay (RREB1, CCND1, MYB, CEP6, 9p21, and CEP9) in discriminating between melanoma and benign nevi is estimated to be approximately 85 % and 95 %, respectively.
While the melanoma FISH assay is utilized as a diagnostic ancillary test in morphologically borderline melanocytic tumors, and is currently available through a number of commercial labs and academic centers, it has its limitations. The accuracy of the test is highly operator dependent, as false positive results can occur due to cherry picking of nuclei, failure to recognize nuclear overlap, and due to the presence of polyploidy. In particular, approximately 5–10 % of Spitz nevi show polyploidy (having multiple the haploid number) [34, 83, 84]. It is also important to note that 9p21 deletions have been described in Spitz and dysplastic nevi but these were heterozygous deletions, in contrast to the homozygous loss seen in melanoma. Therefore, failure to discriminate between heterozygous versus homozygous deletion 9p21 deletion can also result in a false positive result. Finally, a positive result does not equate a diagnosis of melanoma, as this test is an ancillary diagnostic tool, with histopathologic findings still remaining the gold standard. A specific example is the MYB loss in tumors falling into the morphologic category of atypical Spitz tumor. Literature to date suggests a lack of correlation with isolated 6q23 (MYB) loss and aggressive clinical behavior in atypical Spitz tumors, as these appear to behave in an indolent fashion [67, 68].
A negative FISH result in a severely atypical lesion in which the consensus histopathologic assessment is concerning for malignancy should also be interpreted with caution due to possibility of a false negative result (the test has an approximately 15 % false negative rate). False negative results may be due to sampling error, sample handling, aberrations below the level of detection, or aberrations in chromosomal areas other than those tested in the assay. Sensitivity tends to be lowest in melanomas from intermittently sun-damaged skin and spitzoid melanomas (closer to 85 %) [85, 86]. Sensitivity is very low (~50 %) in desmoplastic melanomas, and therefore, these should not generally be submitted for FISH.
Due to the intricacy of the assay itself as well as the need for a clear understanding of the significance of specific aberrations in particular melanocytic tumor subtypes, the melanoma FISH is best done by a cytogeneticist in conjunction with a board certified dermatopathologist familiar with the test [84, 85].
Next-Generation Sequencing Analysis
Next-generation sequencing (NGS) is also used in the molecular analysis of melanocytic tumors. Though NGS requires an elaborate infrastructure for data storage and considerable expertise is required for interpretation, it is becoming increasingly competitive with regard to cost-effectiveness when compared to CGH and FISH [87, 88]. Currently, a number of both commercial laboratories and academic centers offer NGS-based testing for gene panel mutational testing mainly for treatment purposes. The sensitivity of NGS as compared to Sanger sequencing is higher (2–10 % versus 15–25 % allele frequency detection), and less input DNA is needed [36].
Cutaneous Soft Tissue Tumors
Soft tissue tumors are frequently encountered by the practicing dermatopathologist. Although the majority of the most common types of mesenchymal tumors that occur in the dermis and superficial subcutis are benign, sarcomas and soft tissue tumors of intermediate malignancy are also sometimes encountered, and can be diagnostically challenging. Sarcomas and soft tissue tumors in general can be divided into those without recurrent genomic aberrations and those with recurrent typically isolated genetic aberrations (usually either amplifications, translocations or point mutations). Given these types of genetic changes, molecular testing in cutaneous soft tissue tumors typically involves either fluorescence in situ hybridization (FISH) or reverse transcription polymerase chain reaction (RT-PCR) to detect translocations or fusion gene transcripts, or sequencing analysis to identify point mutations. A detailed review of all possible soft tissue tumors that may affect the skin for which molecular testing can play a role is beyond the scope of this chapter, although a list of the more frequently encountered soft tissue tumors with recurrent aberrations is listed in Table 17.5. Three of the more frequently encountered sarcomas with recurrent genetic aberrations that may occur in the dermis and/or superficial subcutis, namely dermatofibrosarcoma protuberans, epithelioid sarcoma, and clear cell sarcoma, are the focus of the following discussion. Further review on soft tissue tumor is in Chap. 13.
Table 17.5
Recurrent genetic aberrations in soft tissue tumors that may occur in the dermis and/or superficial subcutis
Tumor | Recurrent genetic abnormality | Genes involved |
|---|---|---|
Angiomatoid fibrous histiocytoma | t(2;22)(q33;q12.2) t(12;16)(q13;p11) | EWSR1-CREB1 FUS-ATF1 |
Clear cell sarcoma | t(12;22)(q13;q12) t(2;22)(q34;q12) | EWSR1-ATF1 EWSR1-CREB1 |
Dermatofibrosarcoma protuberans | t(17;22)(q22;q13) | COL1A1-PDGFB |
Epithelioid hemangioendothelioma | t(1;3)(p36;q25) | WWTR1-CAMTA1 |
Epithelioid sarcoma | Aberrations involving 22q11 | SMARCB1 (INI1) |
Ewing sarcoma/peripheral primitive neuroectodermal tumor | t(11;22)(q24;q12) t(21;22)(q22;q12) t(7;22)(p22;q12) t(17;22)(q12;q12) t(2;22)(q33;q12) | EWSR1-FLI1 EWSR1-ERG EWSR1-ETV1 EWSR1-E1AF EWSR1-FEV |
Extraskeletal myxoid chondrosarcoma | t(9;22)(q22;q12) t(9;17)(q22;q11) t(9;15)(q22;q21) | EWSR1-NR4A3 RBP56-NR4A3 TCF12-NR4A3 |
Giant cell tumor of tendon sheath | t(1;2)(p13;q35) | COL6A3-CSF1 |
Low grade fibromyxoid sarcoma | t(7;16)(q33;p11) t(11;16)(p11;p11) | FUS-CREB3L2 FUS-CREB3L1 |
Inflammatory myofibroblastic tumor | t(1;2)(q22;p23) t(2;19)(p23;p13) t(2;17)(p23;q23) t(2;2)(p23;q13) | TPM3-ALK TPM4-ALK CLTC-ALK RANBP2-ALK |
Myoepithelial carcinomas of soft tissue | t(19;22)(q13;q12) t(1;22)(q23;q12) t(6;22)(p22;q12) | EWSR1-ZNF444 EWSR1-PBX1 EWSR1-POU5F1 |
Myxoid liposarcoma | t(12;16)(q13;p11) t(12;22)(q13;q12) | FUS-DDIT3 EWSR1-DDIT3 |
Myxoinflammatory fibroblastic sarcoma | t(1;10)(p22;q24) Amplification 3p11-12 | TGFBR3-MGEA5 VGLL3 |
Synovial sarcoma | t(X;18)(p11;q11) | SYT-SSX1 SYT-SSX2 SYT-SSX4 |
Well-differentiated liposarcoma/atypical lipomatous tumor | 12q14-15 amplification | MDM2 CDK4 SAS HMGA2 |
Dermatofibrosarcoma Protuberans
Dermatofibrosarcoma Protuberans (DFSP) is a fibroblastic spindle cell tumor of intermediate malignant potential, which shows a highly infiltrative pattern of growth in the dermis and subcutis (Fig. 17.6). It has a high rate of local recurrence; however, distant metastasis is rare. It typically presents as either a multinodular or plaque-like firm tumor without significant epidermal changes. It is characterized by diffuse strong positivity for CD34 on immunohistochemical staining, and also for a recurrent genetic aberration involving chromosomes 17 and 22. This is either a reciprocal translocation t(17;22)(q22;q13) or a supernumerary ring chromosome which forms from this same translocation, both of which create a fusion gene between collagen type I alpha 1 (COLIA1) on chromosome 17 with platelet-derived growth factor beta-chain gene (PDGFB) on chromosome 22 [89]. This fusion is present in all variants of DFSP, including the giant cell fibroblastoma variant as well as pigmented (Bednar tumor), myxoid, and fibrosarcomatous variants. Therefore, detection of this translocation can aid in the diagnosis of DFSP, as several of these variants can be challenging to diagnose, particularly in a partial biopsy [90]. Confirmation of the translocation also plays a role in treatment, as there are reports efficacy and ongoing clinical trials using in tyrosine kinase inhibitors such as imatinib mesylate in the treatment of advanced, metastatic, and/or unresectable DFSP [91, 92]. Detection of the COLIA1-PDGFB rearrangement is typically via FISH probes specific for the two loci or by RT-PCR. Fresh tissue works best for detection of the transcript via RT-PCR, although the fusion can be detected by RT-PCR in formalin-fixed paraffin-embedded (FFPE) tissue in ~85 % of cases [93].
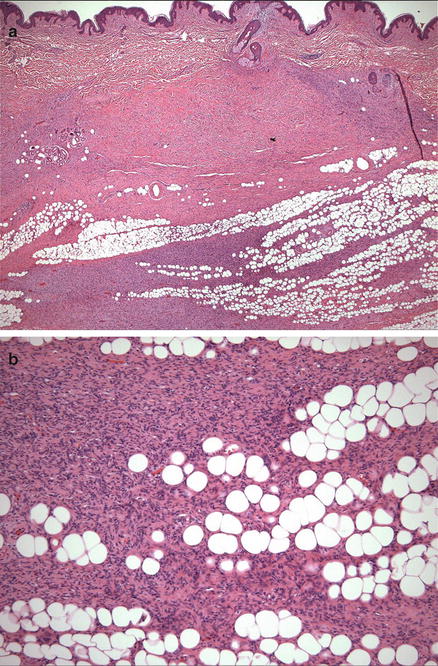
Fig. 17.6
(a) Dermatofibrosarcoma protuberans with infiltration of bland spindle cells into the subcutis. (b) The tumor cells are highly infiltrative into the subcutaneous adipose tissue, with entrapment of fat cells in a “lace-like” or “honeycomb” pattern
Epithelioid Sarcoma
Epithelioid sarcoma is an uncommon sarcoma which tends to occur in young adults in their second to fourth decades of life, with a high rate of recurrence and metastasis [94, 95]. There are two types, namely conventional or distal-type and an even more aggressive clinicopathologic variant known as proximal (large cell) type. Epithelioid sarcoma in general is considered a high grade sarcoma and is treated by wide local surgical excision which can be challenging as distal-type epithelioid sarcoma tends to occur in the distal extremities (particularly the hands, forearms, knees and lower legs) and as proximal-type epithelioid sarcoma has a predilection for the pelvic, perineal and genital areas. Histopathologically, distal-type epithelioid sarcoma grows as a poorly circumscribed ill-defined proliferation of epithelioid to focally spindle cells with vesicular nuclei and small nucleoli often surrounding areas of necrosis, causing a pseudogranulomatous appearance that can be mistaken diagnostically at low power for an inflammatory or infectious process. Vascular invasion is relatively common, although the mitotic rate is typically low (<10 per hpf). In contrast, proximal-type epithelioid sarcoma is composed of markedly atypical enlarged epithelioid cells with vesicular nuclei, and prominent nucleoli. While necrosis is also common in this type as well, it usually lacks the granulomatous quality of the distal-type variant.
Both distal- and proximal-type epithelioid sarcoma show the same immunohistochemical profile, namely diffuse expression of epithelial membrane antigen (EMA) and/or keratins, and approximately 90–95 % of all epithelioid sarcomas show loss of INI1 gene expression by immunohistochemistry [96, 97]. Approximately 50 % of epithelioid sarcomas are also reactive for CD34 by immunohistochemistry.
Aberrations involving the long arm of chromosome 22 (22q) have been identified in both types of epithelioid sarcoma, leading to inactivation of the SMARCB1 (INI1 or hSNF5) gene on chromosome 22 (22q11.2) which is typically either by chromosomal deletion or by point mutation [98–100]. However, detection of INI1 gene deletion and/or point mutation is quite variable (20–50 %) in the literature, suggesting that other genes on chromosome 22 may also be involved [98, 99, 101, 102]. Although alterations in the SMARCB1 locus can be assessed by either conventional FISH, interphase or metaphase FISH, or comparative genomic hybridization, due to the heterogeneity of the inactivating alterations that can occur at this locus (i.e., missense or frameshift mutations in the genes coding region) the most sensitive method is via sequencing analysis of the entire gene.
Clear Cell Sarcoma
Clear cell sarcoma (CCS) is a rare melanin-producing sarcoma which, although sharing some morphologic similarities and positivity for melanocytic markers via immunohistochemistry, is genetically and clinically distinct from cutaneous melanoma. In contrast to cutaneous melanoma, it arises in the deep soft tissue with only rare cases involving the dermis or superficial subcutis. Similarly to epithelioid sarcoma it tends to affect young adults between the ages of 20 and 40. Clear cell sarcoma most commonly arises in the foot or ankle, followed by the knee, thigh, and hands [103, 104]. The tumor is aggressive, with a poor long-term prognosis [103]. Early detection (small tumor size) and wide local excision are required for both local control and a better overall prognosis. Unlike other sarcoma subtypes which tend to spread hematogenously, CCS (and epithelioid sarcoma) tends to metastasize via lymphatics to lymph nodes as well as to visceral sites and bone. Histologically, CCS is composed of epithelioid to spindled cells with clear to slightly eosinophilic cytoplasm, vesicular chromatin, and prominent nucleoli. The cells are disposed in nests or short fascicles often separated by dense fibrous septa and scattered multinucleated cells may be present. Melanin pigment may also be present, although it is typical focal. The cells are fairly monomorphic without significant pleomorphism, and mitoses are low (Fig. 17.7).
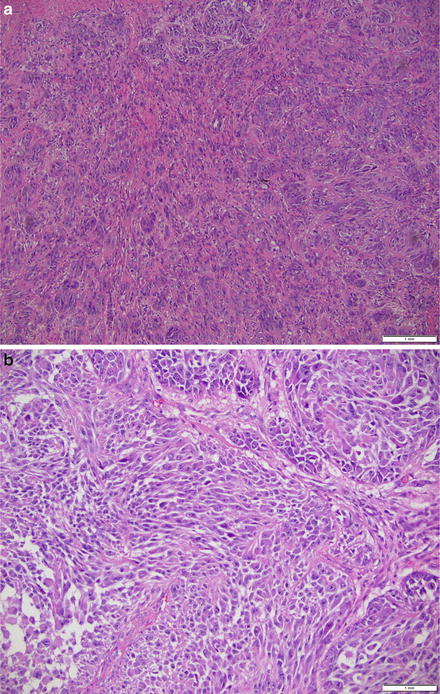
Fig. 17.7
(a) Clear cell sarcoma on the forearm of a 35-year-old arranged in nests of epithelioid to spindled cells with uniform atypia, vesicular chromatin, prominent nucleoli and abundant eosinophilic to amphophilic cytoplasm. (b) Clear cell sarcoma on the forearm of a 35-year-old arranged in nests of epithelioid to spindled cells with uniform atypia, vesicular chromatin, prominent nucleoli, and abundant eosinophilic to amphophilic cytoplasm, at higher magnification
Immunohistochemically, CCS tumor cells express melanocytic markers, including S100, Melan-A/MART-1, microphthalmia transcription factor (MITF), and HMB-45. Interestingly and unlike cutaneous melanoma, HMB-45 staining is usually stronger and more diffuse than S100 staining in CCS. CCS is negative for keratins.
Genetically, clear cell sarcoma is characterized by a reciprocal translocation between chromosomes 12 and 22, t(12;22)(q13;q12) which results in a fusion gene product involving the EWSR1–ATF1 genes [105–107]. This fusion oncogene functions as a transcriptional regulator that causes activation and expression of ATF1 target genes, one of which is MITF, which may at least in part, explain the melanoma-like gene expression profile of CCS [108]. While approximately 90 % of CCS harbor the t(12;22)(q13;q12) translocation, a t(2;22)(q33;q12) translocation fusing the EWS and CREB1 genes is seen in <10 % of CCS [109, 110].
Clear cell sarcoma can be particularly difficult to diagnose and distinguish from melanoma, particularly metastatic melanoma involving the soft tissue as they can appear morphologically and immunophenotypically indistinguishable. Delineation between these two diagnoses is important both for treatment and prognosis. Therefore, detection of the reciprocal translocation can be essential for the accurate diagnosis of CCS. If fresh tissue is available, RT-PCR is the most sensitive method to utilize for detection of the translocation fusion gene product. However, the sensitivity is lower in FFPE tissue (<85 %), and therefore, interphase FISH is a more sensitive approach to use when only FFPE tissue is available for analysis [109].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


