Fig. 5.1
Sanger sequencing traces (a) using the Forward primer, and (b) the Reverse primer. The position of the variant is indicated with an arrow
In addition, the limit of detection of SNVs using Sanger sequencing has been determined to be 15–20 % [6], which may not suffice to detect somatic mutations in fixed tumor specimens.
Next-Generation Sequencing
When compared with Sanger sequencing, the major advantage offered by NGS is the ability to produce an enormous volume of data [7] at a reduced cost per base. This particular feature has expanded the applications of DNA sequencing to allow the resequencing of human genomes in a cost-effective manner. NGS, as opposed to Sanger sequencing, is based on the clonal sequencing of all the nucleic acid molecules present in the sample.
NGS technology can be achieved in a variety of commercial platforms, but they all include the following steps or methods: library and template preparation, sequencing, and data analysis. The library and template preparation step generally involves randomly breaking genomic DNA into smaller sizes or using PCR to amplify short genomic regions of interest within a sample. These short DNA fragments are then typically attached or immobilized to a solid surface or bead. The immobilization of spatially separated short DNA fragments allows millions of sequencing reactions to be performed simultaneously. The clonal amplification of templates can be achieved by solid-phase amplification [8] or by emulsion PCR (emPCR) [9]. Currently, there are several instruments being used in clinical settings that employ either solid-phase amplification or emPCR, while novel technologies are still in a nascent state. During emPCR, for example, after the successful amplification and enrichment of emPCR beads, millions are then immobilized on a chip, manufactured on wafers [10], containing millions of microscopic wells, each one designed to hold only one bead. The sequencing reaction then occurs and the addition of each new base can be detected by the release of a H+. On the other hand, those instruments using solid-phase amplification base the detection of the sequencing reaction by the emission of fluorescence from fluorescently labeled dNTPs. Due to the clonal nature of the NGS technology, the result is a population of template molecules, each of which has undergone the sequencing reaction multiple times. The number of times a molecule is sequenced is represented by the number of reads obtained at the completion of the sequencing run. Such reads are then aligned to a known reference sequence, such as the assembly of the human genome from February 2009 (hg19, GRCh37 Genome Reference Consortium Human Reference 37), which is the most utilized nowadays. This leads to the subsequent identification of variants, or bases that differ from the reference genome used in the alignment process. The number of reads covering a particular base in the genomic sequence is referred as the depth of coverage for that base. Typically, one can obtain a short sequence reads of up to 300–400 bases, with a depth of coverage of 30×, or up to 1,000×, depending on the application. Visualization of the raw alignments generated by this technology can be assessed by using publicly available tools, such as the Broad’s Integrative Genomics Viewer (IGV) [11] (Fig. 5.2). Much higher depth of coverage may be needed for more sensitive applications, such as detecting cell-free circulating tumor DNA in liquid biopsies, which is known as ultra-deep sequencing.
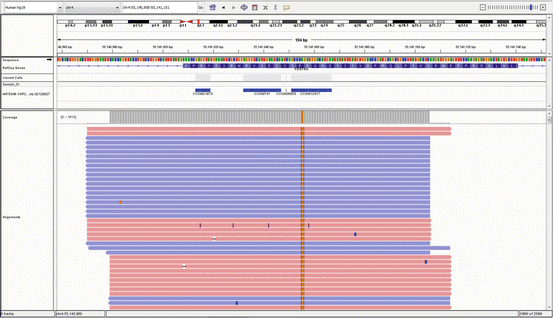
Fig. 5.2
Integrative Genomics Viewer (IGV) display of aligned reads against the hg19 reference sequence, for a defined genomic region of a sample showing a SNV (in orange)
When applying NGS-based assays to variant identification in solid tumor specimens, one must take into account the fact that DNA isolated from fixed samples can be degraded and cross-linked due to formalin fixation, mixed with DNA from nonmalignant cells, or from non-mutant tumor cells, due to tumor heterogeneity, and of very small quantity, due to the increase use of less-invasive procedures, such as fine-needle aspiration (FNA) procedures. Therefore, somatic base calling can be a complex process because the mutant allelic fraction can range between 0 and 1, rather than the fixed 0.5 or 1 expected germline allelic fraction for heterozygous or homozygous mutant, respectively. Thus, in order to be able to detect 5 % mutant alleles, one would need a minimum depth of coverage of 400× [12], which is easily attainable with current NGS platforms.
One of the features of NGS that aide in reducing the cost per base is the fact that multiple samples can be sequenced simultaneously in a single run or reaction by virtue of “barcoding”. Unique sequence tags, or “barcodes” are incorporated into individual patient specimen-derived DNA during the library preparation process. After the sequencing reaction is completed, each individual sample can be sorted out during the bioinformatics pipeline. Such pipeline will align or map the reads of each individual patient sample to the human reference genome sequence. This leads to the subsequent identification of variants, or bases that differ from the reference genome used in the alignment process. A variety of different algorithms have been developed to perform the alignment and the variant calling process, with most of them being platform-specific. Finally, identified variants are further annotated by querying a variety of publicly available databases [13] to provide meaningful reports with clinically relevant information [14] (Fig. 5.3).
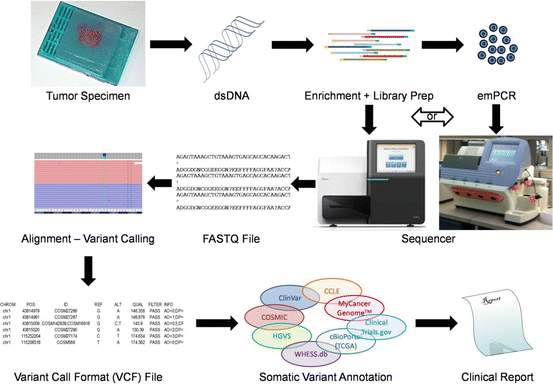
Fig. 5.3
Schematic representation of targeted NGS and annotation for tumor specimens. Double-stranded DNA (dsDNA) is extracted from FFPE specimens containing tumor cells. Targeted regions are enriched by PCR and clonally amplified by either solid-phase amplification on the sequencer or by emulsion PCR (emPCR). Such DNA fragments are sequenced by an NGS instrument, which generate the raw sequence with its corresponding quality Phred scores (FASTQ). Following alignment to a reference genome, variations from such reference are called by variant caller algorithms generating a VCF file. Rich variant annotation for somatic mutations can be achieved by querying multiple publicly available databases to create a meaningful clinical report for the management of oncology patients
Detection of Known Variants
It is noteworthy to differentiate the detection of any mutation in a genomic region or gene with assays design to identify particular mutations of mutations that have previously been defined. Even though one could use the DNA sequencing methods described above, the detection of defined mutations can be achieved by alternative methodologies that are often faster and operate at lowers costs than DNA sequencing. The major limitation of such approaches is that they will only detect the intended mutation. Our knowledge of somatic and actionable variants in solid tumors in particular and in cancer in general is actively growing, and the need to identify multiple mutations in a single assay in small samples is becoming common practice. However, some laboratories may decide to focus their testing to a manageable number of fast mutation detection assays, at low cost. Such methods of detection are discussed in the following sections.
Typically, such methods are based on PCR applications, where primers, and sometimes probes, are designed to specifically detect the sequence of interest within the human genome. The challenge resides in the assay design to ensure sequence specificity for the detection of the intended variant and to avoid nearby polymorphisms that can prevent primer or probe hybridization resulting in no amplification or a false negative result. Such phenomenon is described as “allele dropout”, which happens when some alleles do not amplify but internal controls do. During the assay design process, one can research the sequence of interest for the presence of know single nucleotide polymorphisms (SNPs) in publicly available databases, such as the National Center for Biotechnology Information (NCBI) Short Genetic Variations database (dbSNP) [15].
Real-Time PCR
Real-time PCR, as opposed to end-point or traditional PCR, provides the ability to detect the amplification product as it is generated, by a thermocycler coupled to a constant source of light, e.g., laser or tungsten lamp, and a fluorescence detector, e.g., charge-coupled device (CCD) camera. Real-time PCR has been typically used in quantitative assays, also known as qPCR; however this technology has been successfully applied for genotyping assays, which are qualitative tests. When applied to quantitative assays, one can perform absolute quantitation [16], by using internal or external quantification calibrators; or relative quantitation, by means of detecting a housekeeping [17, 18] or reference gene within the sample, relative to a standard [19].
Several different chemistries have been developed over the years for real-time detection of target sequences [20]. However, the most commonly used platforms nowadays involve the use of hydrolysis probes [21], hybridization probes [22] or Scorpion primers [23]. These three different technologies have been successfully applied to genotyping assays such as allelic discrimination, melting curve analysis, or amplified refractory mutation systems (ARMS), respectively.
These three different chemistries are based on the concept of Fluorescence Resonance Energy Transfer (FRET) [24], referred as the transfer of excited-state energy between two fluorophores, a donor and an acceptor. A fluorophore is a molecule that is capable of rising to an excited state when it absorbs energy from an external source of light, and the process of returning to the basal state results in the emission of energy as fluorescence. When a donor fluorophore is excited, the light emitted by such fluorophore has a lower energy and frequency and a longer wavelength than the absorbed light, and can be transferred to an acceptor fluorophore. When both fluorophores are in close proximity, between 10 and 100 Å [25], then FRET occurs. Depending on the nature of the acceptor and how the energy transferred to that molecule is dissipated, two different FRET mechanisms can be delineated: (1) FRET-based fluorescence, where the transferred energy is emitted as fluorescence due to the fact that the acceptor is also a fluorophore, and (2) FRET-quenching [26] where the electronic energy of the quencher (a nonfluorescent molecule) is dissipated as heat. When choosing a chemistry platform for real-time detection of target sequences careful selection of the quencher/fluorophore pair is critical, since the detection of a positive signal differentiates between the quenched state and the fluorescence of the probe. This can be achieved by many instruments currently available, most of which can discriminate among more than six fluorophores.
One of the most widely employed methods for real-time PCR is the one that uses hydrolysis probes, which relies on the 5′–3′ nuclease activity on the Thermus aquaticus (Taq) DNA polymerase. The use of a dually labeled, target-specific probe confers additional specificity for detection of the amplicon to the reaction. Such probes anneal to the target sequence during the extension phase of the PCR when the nuclease activity of the Taq polymerase removes the fluorophore, releasing it from the probe sequence and removing it from the proximity of the quencher, allowing the detection of the fluorescence, which accumulates with the same kinetics of the amplicon. In some instances, one can design probes with minor groove binding (MGB) ligands to increase the DNA specificity of the probe, allowing the use of short oligonucleotides in allelic-discrimination applications. MGB ligands are small molecule tripeptides, including dihydrocyclopyrroloindole tripeptide (DIP) or 1, 2-dihydro-(3H)-pyrrolo [3.2-e] indole-7-carboxylate (CDPI) that form a non-covalent union with the minor groove of double stranded DNA (dsDNA) [27].
The use of hybridization probes, on the other hand, allows the detection of a fluorescent signal when binding to the DNA target during the annealing phase of PCR amplification. This system allows melting curve analysis to be performed. This chemistry platform consists of a pair of oligonucleotides binding to adjacent target DNA sequences. One probe carries a reporter fluorophore at its 3′-end and the other contains a quencher at its 5′-end and a phosphate group attached to its 3′-end to prevent DNA amplification [28].
More recently, the use of hairpin-loop primer-probes, such as Scorpions, has been adopted for genotyping applications. The hairpin structure contains a reporter at the 5′-end and an internal quencher at the 3′-end. The 3′-end of the hairpin is attached to the 5′-end of the primer by a HEG (hexathylene glycol) blocker to prevent primer extension by the DNA polymerase [23], whereas the loop sequence is designed to match the targeted genomic sequence.
In general, the advantage of real-time PCR is the ability to detect amplicons without further handling of the PCR product, thus minimizing the risk of amplicon contamination. This method can be applied to multiplex reactions through the use of different fluorophores, allowing the detection of multiple genotypes in a single reaction. In addition, the fact that the signal only derives from the target genotype, reduces the noise derived from normal or wild-type alleles when detecting somatic variants in DNA samples from heterogeneous tumor specimens, thus allowing for very low limit of detection (LoD) of such assays.
Allele-Specific PCR
ASPCR, also known as amplification refractory mutation system (ARMS), refers to a PCR amplification where one of the primers, usually the forward primer, extends only when the 3′-terminal base of the primer forms a perfect match with the target sequence. However, the ability of Taq DNA polymerase to prevent extension from a mispaired 3′-terminal base is not absolute, therefore some mispairs may allow extension or mispriming. The application of ASPCR in genotyping or somatic variant detection relies on the primer design, where the last base of the forward primer matches the target mutated base. Thus, the presence of amplicon will indicate the presence of the mutated sequence of interest. This concept can be problematic, since a negative result could correlate with a normal or wild type sequence in the tested sample or the lack of amplification due to the presence of PCR inhibitors, or highly degraded DNA. Thus, it is imperative to include a control reaction, which can be performed on an independent tube or in the same reaction if multiplexing a primer for a conserved base near the target sequence, to ensure that the individual sample DNA is properly amplifiable and avoid false-negative results.
The LoD or analytical sensitivity of ASPCR assays can be limited by the extension of the allele-specific mutant primer on template DNA with the normal sequence, or mispriming. Thus, optimization of the reaction is required during the assay design phase, mainly by titrating primer and magnesium concentrations, in samples containing mixtures of mutated and normal DNA sequences, in different ratios. Depending on the detection method, ASPCR assays have a LoD of one mutated sequence in greater than 100-fold excess of normal DNA, and can be multiplexed as several ARMS assays in one tube, when using fluorescent primers, such as Scorpions (Fig. 5.4).
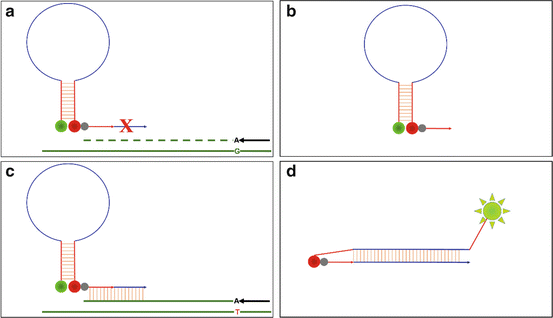
Fig. 5.4
Schematic representation of Scorpion primer used in ARMS assays, where amplification directed by the allele-specific primer, which has an A at the 3′ end, is (a) prevented in the presence of the wild-type base, G, resulting in (b) a stable hairpin-loop conformation and no signal from the Scorpion; or (c) amplification is enabled in the presence of the mutant base, T, thus generating the template for the Scorpion primer to bind, elongate, and create a complementary sequence to the loop—blue line, causing (d) the disruption of the hairpin conformation releasing the reporter (green circle) from the proximity of the FRET-quencher (red circle), and emit fluorescent light
Single-Base Primer Extension
Single-base primer extension is an application based on the specificity of the Taq DNA polymerase that can be successfully applied to the detection of somatic SNVs in tumor samples. The features of single-base primer extension consist of a PCR performed using primers flanking the target variant site, followed by a second reaction, similar to Sanger sequencing, where fluorescently labeled ddNTPs, complementary to the target base, are incorporated at the 3′-end of primers that are designed to bind to the amplicon immediately adjacent to the base being interrogated [29]. The resulting labeled primers can then be detected by capillary electrophoresis. Thus, the extended primers are identified by the combination of the fluorescent dye of the incorporated ddNTP and the size of the terminated products. Multiplexing can be achieved by using different sizes of the extended primers designed to detect several separate genotypes within the same or different amplicons in the same reaction, where 5–10 variants can easily be discriminated, based on size and/or color.
This method has been successfully applied to the detection of germline SNVs, but its LoD, down to 5 %, allows for the sensitive detection of somatic SNVs in solid tumors. This technology can be easily applied to short DNA amplicons (100–200 bp) making it ideal for DNA isolated from formalin-fixed, paraffin-embedded (FFPE) tissue samples.
Detection of Unknown Variants
Molecular methods for the identification of somatic mutations are often needed in situations where there the target is not a unique SNV, but a variety of different SNVs within a codon or exon that have similar diagnostic or predictive value. In such scenarios, the molecular assay should be designed to detect potentially unknown variants within a genomic region. This can be achieved by the detection of heteroduplex formations during the hybridization of partly complementary DNA strands, or by DNA sequencing methods.
High-Resolution Melting Analysis
One method to detect heteroduplex formations during the hybridization of partly complementary DNA strands, is by melting curve analysis. Nonspecific intercalating dyes that fluoresce while binding to dsDNA [30] can be used for High-Resolution Melting Analysis (HRMA) to rapidly screen a defined genomic region for the presence of mutations. Thus, PCR is carried out using primers flanking the genomic region of interest in a reaction mix containing the dye. After amplicon generation, the reaction is gradually heated from 50 to 95 °C, causing the dsDNA to melt, thus decreasing the amount of fluorescence. The temperature corresponding to half of the DNA molecules being melted is defined as the melting point, or T m, and is the inflexion point in the melting curve produced by plotting amount of fluorescence versus temperature. The amplification of DNA forms heterogeneous samples containing both normal and mutated DNA sequences, such as solid tumors samples, will yield a mixture of homoduplex and heteroduplex amplicons. Since less energy is required to break a lesser number of hydrogen bonds in heteroduplex amplicons, these will have a cooler T m compared to the one from homoduplex amplicons, resulting in a melting curve with two inflexion points. In the unlikely event that the patient specimen consists entirely of homozygous mutant neoplastic cells this method will fail to distinguish the T m from a wild type homoduplex. Alternatively, one can use fluorescently labeled hybridization probes instead of nonspecific intercalating dyes to allow for the distinction between different T m [31].
Even with the use of hybridization probes, HRMA is nonspecific, since DNA polymorphisms can lead to the changes in T m, thus requiring follow-up confirmation using an alternative methodology, such as DNA sequencing, to determine the nature of the exact DNA variant. Typically, the LoD for mutations using HRMA has been reported to around 10 %, but, in general, it should be carefully determined by each individual laboratory for its HRMA assays.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


