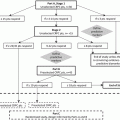
Phenotype
Molecular basis
Mechanism
Consequence
AR-addicted
Adrenal or intra-tumoral androgen biosynthesis
Androgens produced sufficient to activate AR
Responds to androgen biosynthesis inhibitors and selected AR antagonists
AR mutation
Alters ligand specificity
Increases ligand promiscuity
Triggers antagonist-to-agonist switch
May respond to selected AR antagonists
AR over-expression
Increases ligand sensitivity
Increases ligand promiscuity
Often responds to selected AR antagonists
AR splice variant
Provides ligand-independent receptor function
Will not respond to AR antagonists
AR post-translational modification
Increases receptor activity
May respond to AR antagonists
Non-AR-addicted (AR pathway-independent prostate cancer or APIPC)
Neuroendocrine prostate cancer (NEPC)
Driven by N-MYC, AURKA, and other genes
Will not respond to androgen biosynthesis inhibitors or AR antagonists
Other
Not known
Will not respond to androgen biosynthesis inhibitors or AR antagonists
The Molecular Biology of AR Function
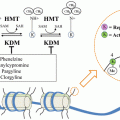
AR is a ligand-dependent transcription factor encoded by a single gene with 8 exons located at Xq11-12. The receptor is a member of the nuclear receptor superfamily group that also contains the glucocorticoid, mineralocorticoid, and progesterone receptors 3 [17]. The physiologic AR protein shows marked inter-individual differences in size as a result of variable polyglutamine and polyglycine repeats. These differences may affect receptor function, with both increased transcriptional trans-activation and increased prostate cancer risk seen in association with AR containing shorter polyglutamine repeats [18, 19]. AR structure can be considered in terms of mapped functional domains, including a ligand binding domain (LBD) ensuring selective activation by androgenic hormones, a hinge domain, a DNA binding domain (DBD) permitting binding selective binding to ARE sequences, and an N-terminal domain; critical regions for transcription trans-activation (activation function or AF regions) are located both in the LBD and in the N-terminal region [1]. In the absence of androgens, the receptor is sequestered in the cytoplasm via an interaction with chaperone proteins. Hormone binding triggers a cascade of events starting with a change in AR conformation which results in liberation from chaperones, dimerization, and ingress into the cell nucleus [1].
The arrival of the ligand-bound, activated, AR in the cell nucleus attracts a myriad of transcriptional co-regulators, including histone acetyltransferases, histone demethylases, SWI/SNF proteins, poly(ADP-ribose) polymerase (PARP), and as mentioned, TOP2B, along with as many as 200 or more other proteins represented in several complexes [1, 14, 20, 21]. These complexes then act to modify chromatin proteins so as to sculpt an active chromatin conformation capable of loading RNA polymerase II at target genes. The activated AR is competent to directly engage co-regulatory proteins containing an FxxLF amino acid motif upon ligand binding as a result of the movement of helix 12 in the LBD to create a hydrophobic pocket (AF-2 [22]). The AR itself has an FQNLF amino acid sequence within its N-terminal domain, and androgen binding to the receptor can trigger a dimeric N-terminal to C-terminal conformation by virtue of interactions of FQNLF with the unveiled hydrophobic region in the LBD [20]. AR complexes bind genomic DNA not only at ARE sequences located in proximal promoters of genes but also at enhancer elements located far upstream, in introns, and in 3’ untranslated regions [23]. Once in the cell nucleus, activated AR can also modulate a number of genome functions, including facilitating the activation or repression of other genes and promoting licensing of DNA replication origins [24]. Of note, ligand-bound, activated, AR remaining in the cytoplasm has been reported to interact with kinases such as SRC to initiate additional signaling programs [25]. The transcription output attributable to activated AR in normal prostate cells includes differentiation genes such as KLK2, KLK3, and TMPRSS2, while in CRPC cells, AR also tends to promote expression of cell-cycle genes such as CDC20, UBE2C, CDK1, and ANAPC10 [23].
Molecular Mechanisms of Maintained AR Addiction in Many Cases of CRPC
Despite frequent initial beneficial treatment responses to androgen deprivation therapies that lower circulating testosterone levels to <50 ng/mL, CRPC tends eventually to emerge and progress to ultimately threaten life. Intriguingly, CRPC is most often heralded by progressive rises in serum PSA, a biomarker requiring activated AR function in prostate cancer cells [26]. This implies that there is ongoing AR signaling in these cancers, hinting they likely have remained addicted to AR [5]. How does this addiction persist? One mechanism involves ongoing production of androgens, either by adrenals or by the cancer itself, which persists despite ablation of testicular androgen biosynthesis [27, 28]. At cancer sites, T and DHT can be produced at levels sufficient to activate AR either by conversion of adrenal androgen precursors or by new synthesis using CYP17 [27, 28]. This process can be antagonized by ketoconazole, an antifungal drug capable of inhibiting CYP17 when administered at high doses, and by abiraterone acetate, a pregnenolone analog now approved for treatment of CRPC based on survival prolongation in randomized clinical trials [29]. A second CYP17 inhibitor, orteronel (TAK-700), is in advanced clinical development [30].
Another AR-addicted CRPC phenotype can be attributed to new AR mutations, not present at disease presentation, which encode receptors with altered ligand specificity. These cases tend to arise in the setting of treatment with “first-generation” anti-androgens, such as flutamide and bicalutamide, and may be responsible for an “anti-androgen withdrawal” syndrome, where men with disease progression despite the combination of androgen deprivation and anti-androgen administration show improvement upon cessation of anti-androgen treatment but maintenance of androgen deprivation [6, 31, 32]. Such AR mutations do not arise commonly in the setting of androgen deprivation alone. Even in the absence of AR mutations AR-addicted CPRC cases may arise as a consequence of ongoing AR activation stimulated by growth factor signaling pathways capable of creating post-translational modifications of AR or its co-activators [1, 5, 26].
In a study of CRPC arising among several different human prostate cancer xenografts propagated in immunodeficient mice, the most consistent molecular finding was that the abundance of AR was increased, leading to augmented transcriptional trans-activation at lower androgenic hormone levels and in response to a more promiscuous collection of ligands [33]. Amplification of AR has been reported in as many as 80 % of CPRC cancers, with some 30 % exhibiting marked gene amplification, which may account for some instances of AR over-expression [34, 35]. This high AR expression phenotype was exploited in the discovery of the “second-generation” anti-androgens enzalutamide and ARN-509, drugs that appear capable of interfering with AR function despite high-level AR expression in prostate cancer cells that otherwise do not respond to “first-generation” anti-androgen treatment [36, 37]. When compared to the “first-generation” anti-androgens, these new agents exhibit less mixed agonist and antagonist activity when docking to the LBD, triggering a different location of helix 12. The drugs stop AR activation almost entirely, before receptor trafficking to the cell nucleus and binding to ARE sequences [36, 37]. Thus far, enzalutamide has gained approval for CRPC treatment based on randomized trial data showing a survival benefit and ARN-509 trials are ongoing [38].
Predictably, the growing use of enzalutamide for CRPC has fostered the emergence of cancers resistant to “second-generation” anti-androgens. In many such treatment-resistant cases, the AR signaling addiction appears maintained. Model studies of the acquisition of “second-generation” anti-androgen resistance using LNCaP prostate cancer cells have revealed a new mutant AR with an F876L amino acid change in the receptor LBD [39–41]. Enzalutamide was able to bind the F876L-AR with 48-fold greater affinity that wild-type AR; ARN-509 also bound F876L-AR [39]. When the F876L-AR was introduced into prostate cancer cells, enzalutamide and ARN-509 acted as agonists rather than as antagonists, driving AR target gene expression and stimulating prostate cancer cell growth in vitro and in vivo [39, 40]. The F876L-AR mutation has been detected in men with progressive CPRC despite “second-generation” anti-androgen treatment. When plasma DNA from a phase 1 clinical trial of ARN-509 for CPRC was assayed for AR mutations, 3 of 18 men with progressive serum PSA increases despite treatment were found to have a C to A missense change at AR nucleotide 2628 encoding the F876L-AR [39]. Already, drug discovery efforts are underway for “next-generation” anti-androgens which can inhibit signaling by F876L-AR, raising the possibility that as long as CRPC remains addicted to AR with an intact LBD, small molecule antagonist drug therapy may be feasible [41].
A final phenotype of maintained AR addiction in CRPC may be mediated by AR splice variants that encode receptors without LBDs that can nonetheless act to promote target gene transcription [42–46]. The variant AR transcripts contain deleted or cryptically inserted exons. Some such transcripts may be generated by defective AR genes arising in association with AR amplification or other AR rearrangements [47]. However, the majority of the variant AR transcripts likely arise a result of some sort of perturbation in transcription initiation and elongation rates that accompanies androgen deprivation [48]. The resultant receptors contain truncated C-terminus, with an intact N-terminal domain and DBD, but not a functioning LBD. Usually, in prostate cancer cells, the level of variant AR mRNA tends to be far less than that of full-length AR mRNA. However, this may underestimate the expression level of the truncated AR forms encoded by variant AR transcripts, which may be as high as 30 % or more of AR protein [49]. Forced expression of one such truncated receptor, AR-V7 which lacks an LBD and contains 16 amino acids from a cryptic exon, triggered expression cell-cycle regulatory genes in prostate cancer cells, whether or not androgens were present [50]. This AR variant, which has been detected in men with CRPC, can drive the growth of prostate cancer cells in the absence of androgenic hormones. Also, while both full-length AR and AR-V7 tend to promote expression of prostate differentiation genes like KLK3 and TMPRSS2, there are tantalizing differences, as yet unexplained, in the patterns of genes induced by each receptor [50]. Several of the AR variants so far detected also appear to be expressed in CPRC cells and to possess the propensity to propagate ligand-independent signals, while others may not act in this manner [20]. Nonetheless, data are accumulating to suggest that AR splice variants may contribute both to abiraterone resistance and to enzalutamide resistance in CRPC [51].
CRPC Abandonment of AR Addiction
Progression to CRPC, in part driven by highly unstable genomes and epigenetic regulation and in part driven by therapeutic pressure, can result in a phenotype/genotype that is entirely independent of AR signaling, a tumor state that has recently been operationally termed Androgen Receptor Pathway-Independent Prostate Cancer (APIPC) [52]. One type of APIPC can be characterized by features such as loss of PSA production, lytic bone metastases, hypercalcemia, or widely disseminated visceral metastases that are otherwise uncommon complications of systemically advanced prostate cancer. This prostate cancer cell phenotype, often referred to as neuroendocrine prostate cancer (NEPC), does not manifest AR expression or AR signaling, representing an escape from AR addiction [53]. Molecular “archeology” studies of NEPC strongly suggest that most such cases evolved from AR-addicted prostate cancers, as many have been found to contain TMPRSS2–ERG rearrangements [54]. Despite the presence of the rearrangements, the absence of AR signaling prevents TMPRSS2–ERG fusion mRNA or ERG protein expression. AR-independent NEPC cases appear instead to contain other genome alterations, such as amplification of N-MYC and AURKA [55]. For this reason, the NEPC variant of CRPC does not respond to treatments targeted at the AR signaling axis. Instead, the use of cytotoxic chemotherapy, usually with platinum compounds, is often attempted [56]. Ongoing clinical trials are assessing whether AURKA inhibitors might provide benefit to men with NEPC and AURKA amplification with AURKA over-expression. Of concern, though this AR-independent NEPC variant of CRPC was once thought to be rare, the frequency with which it appears may be increasing with the introduction of androgen biosynthesis inhibitors and “second-generation” anti-androgens [57]. CRPC progression to NEPC is clearly dangerous, autopsy studies of life-threatening CRPC have hinted that as many as 25 % or more of men who die with prostate cancer show signs of NEPC [58].
Recent evidence suggests prostate cancers transitioning from an AR-driven to an APIPC state are not obligated to progress via a neuroendocrine phenotype, but may be dependent on alternative survival pathways. For example, as many as 80 % or more of CRPC cases show increased FGF8 expression, and cases with amplification of FGFR2 have been identified [59, 60]. As men live longer with CRPC by responding to the new androgen biosynthesis inhibitors and better anti-androgens, more subtypes of APIPC are expected to emerge [61].
The Molecular Pathogenesis of Lethal Prostate Cancer: Is the Emergence of CRPC Inevitable?
To explain anti-neoplastic drug resistance, Goldie and Coldman applied principles first elaborated by Luria and Delbrück in classic studies of the resistance of bacteria to phage lysis to discriminate the contributions of spontaneous versus induced mutagenesis [62, 63]. Of course, all human cancer cells arise as a consequence of somatic genome errors, and most tend to show an increased propensity for genome instability. Cancer genome sequencing and other genome analyses have disclosed abundant base changes, insertions, deletions, amplifications, chromosome copy number changes, and DNA methylation differences in prostate cancers and other human cancers [64]. Early estimates hint that prostate cancer genomes contain on the order of 3,866 base changes, 108 rearrangements, and 5,408 differentially hypermethylated sequences [65, 66]. Ongoing genome instability can clearly result in cancer treatment resistance. Studies of cancer cell resistance to anti-metabolites and cytotoxic agents have consistently implicated a spontaneous mutation process of some sort as responsible for the emergence of resistant cancer cell clones among cells that were otherwise sensitive to the drugs. Such spontaneous mutation rates have been reported to be high as 1 in 104 per cell/generation in model studies [67]. For prostate cancer, both spontaneous and induced genome alterations probably contribute to disease progression to castration-resistance. The propensity for AR to recruit TOP2B to the regulatory regions of its target genes in prostate cancer cells and trigger directed chromosomal translocations may be a source of induced genome defects that can drive CRPC [14]. As an example, AR–TOP2B-associated DNA double strand breaks might conceivably promote AR amplification and AR over-expression. Prostate cancers have been proposed to evolve via rare widespread chromosomal rearrangement events termed “chromoplexy,” especially in cases showing AR-regulated fusion genes [68]. In contrast, spontaneous mutagenesis is likely responsible for the appearance of F876L-AR in men with CRPC treated with “second-generation” anti-androgens.
Genome-scale analyses of CRPC recovered at autopsy have consistently implicated a single lethal clone and its progeny as responsible for metastatic dissemination and ultimate life-threatening progression. However, ongoing genome instability in progeny of the lethal clone has been a consistent finding, suggesting that many, if not most, prostate cancers will be able to progress despite currently available treatment with androgen deprivation, inhibition of androgen synthesis, “second-generation” anti-androgens, and taxane chemotherapy, as well as to ultimately evade any future attempt at targeted therapy. In one study, genome copy number analysis showed greater similarity in losses and gains among metastatic cancer deposits in one case versus another, but distinct differences among the metastases in each case [69]. An analysis of somatic DNA hypermethylation changes capable of affecting gene expression delivered a similar result [70]. Intriguingly, in this study loss of cytosine methylation, evident in all metastatic prostate cancers, showed marked differences in every metastatic lesion, even within a single CRPC case, regardless of metastatic site. Hypomethylation appears to vary cell-to-cell in metastatic prostate cancer [67]. In addition to reducing the fidelity of suppression of normally silenced embryonic genes, like NY-ESO and others, this epigenetic instability may augment genetic instability in prostate cancer cells via activating retrotransposons and reducing chromatin barriers to repeat sequence recombination [70, 71].
Conclusions
The initial sensitivity of prostate cancers to androgen deprivation likely reflects a redirection of AR signaling from terminal differentiation toward maintenance of a malignant phenotype. This prostate cancer cell addiction to AR forms the basis for androgen deprivation therapy, the most widely used systemic treatment for advanced prostate cancer. Unfortunately, prostate cancers show enough genetic and epigenetic instability that disease progression to castration-resistance is inevitable. CPRC that has remained addicted to AR, particularly if the AR contains an intact LBD, is often amenable to treatment with androgen synthesis inhibitors and “second-generation” anti-androgens that selectively target the receptor LBD. CRPC that is driven by truncated AR encoded by splice variant AR transcripts, or that has escaped AR addiction, tends to be refractory to such drugs. AR-independent prostate cancers appear to be highly aggressive and difficult to treat with the current armamentarium of available anti-neoplastic drugs.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


