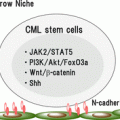
Fig. 2.1
CML stem cell maintenance. CML stem cells self-renew as HSCs do when BCR–ABL kinase activity is inhibited by tyrosine kinase inhibitors (TKIs). Therefore, survival of CML stem cells causes recurrence of CML disease when TKI therapy is discontinued. Survival and self-renewal of CML stem cells despite TKI treatment are supported by several signals that are involved in stemness, the cell cycle, metabolism, epigenetics, or the microenvironment. In addition, molecules downstream of BCR–ABL that function independently of its kinase activity also support the TKI resistance of CML stem cells
Although the molecular mechanisms of CML disease development and progression in patients has been well investigated, it has not been clear whether these mechanisms maintain CML stem cells after the establishment of CML disease or whether CML stem cells are maintained by distinct molecular mechanisms. Because the disruption of signals required for the maintenance of CML stem cells will contribute to the eradication of CML disease, many researchers investigating CML disease have been interested in how CML stem cells are maintained. The maintenance of normal hematopoiesis provides some insight. HSCs are tightly regulated by both intrinsic and extrinsic factors in vivo. Therefore, numerous signals may also be involved in CML stem cell maintenance. For example, accumulating evidence has revealed the presence of “stemness” signals, that is, molecules and signaling pathways that play critical roles in maintaining the undifferentiated properties of various types of stem cells, including embryonic stem cells [5]. Such stemness signals are involved in control of CML stem cell function. Cell cycle regulation may be important, because CML stem cells cycle slowly, like HSCs in vivo, and there is evidence that loss of key cell cycle regulators leads to defective phenotypes in quiescence that are associated with reduced HSC function [6]. Cellular metabolism is also important for the maintenance of HSC function [7]. Furthermore, several microenvironmental components in BM, including the vasculature, endosteal osteolineage cells, and mature hematopoietic cells [8], may affect CML stem cells, although it is unclear whether the CML stem cell niche is identical to that of HSCs. In addition, modulation of metabolism is critical for HSC behaviors [9–12]. Epigenetic modifications are also important for controlling the undifferentiated status of HSCs [13].
Despite the similarities in the regulation of undifferentiated status between CML stem cells and HSCs, there are also critical differences between them. Even if a CML stem cell is derived from a HSC, additional characteristics associated with BCR–ABL are likely acquired during leukemogenesis. Therefore, dissection of BCR–ABL signals, whether or not they depend on its kinase activity, is needed to understand the mechanisms of CML stem cell maintenance. This chapter will introduce recent advances in the understanding of the molecular mechanisms regulating the maintenance of stem cell populations in CML.
2.2 Stemness
Increasing evidence points to shared core molecular stemness mechanisms between normal tissue stem cells and CML stem cells, which creates emerging possibilities for developing new therapeutics for CML. The Wnt and Hedgehog signals are most representative of stemness regulatory pathways. The Wnt proteins are secreted signaling molecules that regulate diverse processes, including embryogenesis, cell polarity, and cell fate modulation of stem/progenitor cells, and their deregulation is observed in many types of cancer [5, 14]. To date, 19 members of the Wnt family have been identified in primate, and these proteins activate two main Wnt signaling pathways, the canonical and noncanonical Wnt cascades. A hallmark of the canonical Wnt pathway is the nuclear localization of β-catenin. When the canonical Wnt signaling pathway is inactivated, β-catenin forms a complex with Axin, adenomatous polyposis coli, and glycogen synthase kinase-3β, and β-catenin is then phosphorylated and targeted for ubiquitinylation and subsequent degradation by proteasomes. Signaling is initiated when Wnt binds to Frizzled receptors and a member of the LDL receptor family, Lrp5/6. In the presence of Wnt signaling, β-catenin is uncoupled from the complex and translocates to the nucleus, where it binds Lef/Tcf transcription factors, thus activating transcription of target genes. The Wnt signaling pathway is critical for many types of tissue stem cells, including epidermal, intestinal, neural, and hematopoietic stem cells [5]. In a mouse model, Wnt3A can induce self-renewal of HSCs and enhance their ability to reconstitute recipient hematopoiesis when transplanted into an irradiated recipient [15, 16]. Furthermore, treatment of undifferentiated human hematopoietic progenitors with Wnt5A leads to their expansion in vitro [17]. In the BM, Wnt proteins are produced by HSCs themselves as well as by the microenvironment, suggesting that Wnt proteins regulate HSCs in both an autocrine manner and paracrine manner [18]. Overexpression of Axin suppresses HSC growth [15], also supporting a pivotal role for the Wnt signaling pathway in the maintenance of HSC stemness. Paradoxically, conditional gene-targeting studies revealed that β-catenin, an essential downstream molecule in the canonical Wnt signaling pathway, is dispensable for normal hematopoiesis and lymphopoiesis [19]. Although the role of β-catenin signaling in the regulation of normal HSCs remains under debate, it is widely accepted that Wnt/β-catenin signaling is necessary for leukemogenesis and development of leukemia initiating cells (LICs) [20–23]. Granulocyte–macrophage progenitors (GMPs) from CML patients with blast crisis show the activation of Wnt/β-catenin signaling, as determined by Wnt reporter activity and accumulation of nuclear β-catenin [24]. Overactivation of Wnt signaling endows GMPs with the stem cell-like property of long-term renewal, which normal GMPs do not possess [24]. Interestingly, after CML was established in a mouse model of BCR–ABL-induced CML, deletion of β-catenin did not increase survival; however, deletion of β-catenin and cotreatment with imatinib synergized to eliminate CML stem cells [25]. This effect was also seen with pharmacological inhibition of β-catenin with indomethacin. These studies suggest that the Wnt/β-catenin signaling pathway is important for the establishment of stemness in progenitors, and it could become a therapeutic target in CML patients.
The Hedgehog (Hh) pathway is another important regulator of embryogenesis that has also been implicated in the development of multiple types of cancer [26]; it is essential for regulating the proliferation, migration, and differentiation of stem/progenitor cells. Genetic mutations of the Hh pathway are found in familial (Gorlin’s syndrome) and sporadic basal cell carcinoma, as well as medulloblastoma, indicating a clear relationship between Hh pathway activity and oncogenesis [26]. In mammals, three Hh proteins, Desert Hedgehog (DHh), Indian Hedgehog (IHh), and Sonic Hedgehog (SHh), function as ligands for Patched (Ptch1). In the absence of an Hh ligand, the Ptch1 receptor inhibits the action of Smoothened (Smo), a G protein-coupled receptor. Binding of Hh ligands to Ptch1 causes internalization and degradation of Ptch1, releasing its suppression of Smo. Smo then interacts with Suppressor of fused (SUFU), which in turn activates glioma-associated oncogene homologue 1 (GLI1) and GLI2 and degrades GLI3, leading to the transcription of tumor-promoting genes.
The role of Hh signaling in normal hematopoiesis is controversial [27]. Evidence from genetic knockouts suggests that the Hh signaling pathway regulates definitive hematopoiesis during development, rather than early primitive hematopoiesis. Loss of Ihh causes hypoplasia of fetal liver, which is a major fetal hematopoietic organ [28]. In addition, overactivation of Hh signaling by single Ptch1 gene deletion (Ptch1 +/−) induces expansion but exhaustion of regenerating HSCs [29]. In contrast, studies using conditional Smo KO mice revealed that Hh signaling is dispensable for adult HSC function [30, 31]. The role of Hh signaling may be highly context dependent, changing with developmental stage, cell type, and even physiologic condition [27]. However, experimental CML models have suggested that the Hh signaling pathway is essential for the maintenance of CML stemness, and this pathway is therefore a therapeutic target. In the Smo KO CML mice, the CML stem cell population was significantly decreased, and the development of re-transplantable BCR–ABL-positive CML stem cells was abolished by the deletion of Smo genes [32]. In addition, treatment with both cyclopamine (Hh inhibitor) and nilotinib produces an additive effect over nilotinib alone in reducing the total number of progenitors and extending the time to relapse after discontinuation of therapy [33]. Therefore, a clinical strategy incorporating both Hh and BCR–ABL inhibition may have value in preventing the drug resistance and disease recurrence associated with TKI treatment alone.
2.3 Cell Cycle
Several studies have shown that CML stem cells cycle more slowly than progenitor CML cells [6]. The similarity in cell cycle status between normal HSCs and CML stem cells provides important clues about understanding the features of CML. One of the master regulators of the cell cycle in normal HSCs and CML stem cells is Fbxw7 (F-box and WD40 repeat domain-containing 7). Fbxw7 is an F-box protein component of an SCF (Skip-Cul1-F-box protein)-type ubiquitin ligase [34]. Fbxw7 has several substrates for ubiquitination, including c-Myc, cyclin E, and Notch1, and regulates both the cell cycle and cellular differentiation. Fbxw7 has three isoforms (α, β, and γ), and Fbxw7α is exclusively expressed in undifferentiated hematopoietic cells and T cell-committed progenitors. Ablation of Fbxw7 in hematopoietic cells has shown the importance of the Fbxw7/c-Myc axis in HSC maintenance [35, 36]. c-Myc is maintained in HSCs at a low level in the steady state and plays a critical role in the self-renewal and differentiation of HSCs [37]. Elimination of c-Myc increases the number of HSCs that can self-renew but fail to differentiate. Overexpression of c-Myc in HSCs enhances differentiation, resulting in HSC exhaustion. In mice with an Fbxw7 deletion, c-Myc protein accumulates in the HSC population, leading to cell cycle entry and premature loss by p53-dependent apoptosis of HSCs [35, 36]. This defective phenotype of Fbxw7-deficient HSCs is rescued by decreasing c-Myc protein expression [38]. Furthermore, the gene expression profile in Fbxw7-deficient HSCs/progenitor cells showed that differentiation-related genes were activated and genes enriched in HSCs were downregulated. Among genes regulating the cell cycle, loss of Fbxw7 in HSCs induced the upregulation of E2F2 and Ccnd1 and the downregulation of p57kip2. These affected genes have been suggested to be targets of c-Myc [39–41]. Conversely, forced expression of Fbxw7 in HSCs/progenitor cells suppresses the accumulation of c-Myc, resulting in the repression of cell cycle progression and maintenance of the high reconstitution activity of HSCs [42]. Fbxw7-overexpressing HSCs show increased expression levels of p21cip1 and p27kip1. Thus, the Fbxw7/c-Myc axis plays an important role in cell cycle regulation and the maintenance of HSCs [43]. In mouse CML models and human patients, Fbxw7 is highly expressed in the stem cell compartment compared to its expression in progenitor and differentiated cells. Genetic ablation of Fbxw7 in CML cells suppresses the initiation and progression of CML disease [44, 45]. Loss of Fbxw7 induces the accumulation of c-Myc, cell cycle entry, and p53-dependent apoptosis, leading to exhaustion of CML stem cells. These functions in Fbxw7-deficient CML stem cells are rescued by a decrease in the c-Myc protein level or silencing of p53. Furthermore, the Fbxw7-deficient CML stem cells are sensitive to anticancer drug treatments. Importantly, the activation of BCR–ABL induces the upregulation of Fbxw7 expression, and the expression levels of Fbxw7 and c-Myc in CML stem cells are higher than those in normal HSCs. The expression pattern shows that Fbxw7 deficiency creates more dramatic defects in CML stem cells than in HSCs. The difference in cycle phenotypes between CML stem cells and HSCs may explain why Fbxw7-deficient CML stem cells show higher sensitivity to imatinib or Ara-C treatment than Fbxw7-deficient HSCs. Thus, Fbxw7 itself or related signals may be a therapeutic target for CML stem cells in combination with TKI.
FOXO transcription factors belong to the forkhead family of transcriptional regulators [46, 47]. In mammals, the FOXO group contains four members: FOXO1, FOXO3a, FOXO4, and FOXO6. FOXO proteins are normally present in an active state in a cell’s nucleus. In response to growth factors or insulin, phosphatidylinositol 3-kinase (PI3K) is activated. PI3K in turn activates several serine/threonine kinases, including protein kinase B (PKB/Akt) and the related SGK family enzymes. Activated Akt phosphorylates FOXO proteins at three Akt phosphorylation sites, resulting in their export from the nucleus into the cytoplasm and subsequently their inactivation. FOXO activity is regulated by several environmental stimuli through posttranslational modifications, including phosphorylation, acetylation, ubiquitination, and methylation. The FOXO family has numerous target molecules that are involved in variety of cellular responses, including cell cycle arrest, DNA damage response, detoxification of reactive oxygen species (ROS), glucose metabolism, and mitochondrial activity. In normal quiescent HSCs, FOXO3a is localized in nuclei, whereas it is predominantly located in the cytoplasm of cycling progenitor cells [48, 49]. Deficiency of FOXO3a protein leads to impaired quiescence of HSCs and elevated ROS, resulting in defective capacity for hematopoietic regeneration after transplantation of HSCs [49–51]. The inhibition of ROS by N-acetyl cysteine reverses the defective phenotype of HSCs, indicating that FOXO proteins contribute to the maintenance of HSCs by suppressing ROS. In a mouse CML model, FOXO proteins were localized in the nuclei of CML stem cells and were associated with the inactivation of Akt, suggesting that the FOXO family may be activated in the these cells, as they are in normal HSCs [52]. FOXO3a is localized in both the cytoplasm and nuclei of CD34+ cells from human CML patients (CML stem cells/progenitor cells), whereas it is localized mainly in the nuclei of normal CD34+ cells [53]. The inactivation of BCR–ABL by TKIs also activates FOXO by inactivating Akt. TKI treatment causes cell cycle arrest and apoptosis of CML cells that are associated with nuclear localization of FOXO1 and FOXO3a. TKI treatment induces several genes that are mediated by the FOXO family, including ATM, p57, and BCL6. Overexpression of FOXO3a in CML cell lines produces G1 arrest and apoptosis; however, CML stem cells derived from patients show an only slight increase in apoptosis when FOXO3a is activated, whereas remarkable cell cycle arrest occurs. Thus, FOXO family proteins are critical cell cycle regulators in CML stem cells, whereas apoptosis induced by FOXO activation may depend on cell context, and only cells with levels of FOXO activity over an “apoptosis threshold” may die. In human CML cell lines, knockdown of FOXO3a inhibits the cell cycle arrest induced by TKI treatment, followed by the induction of cell death. Importantly, FOXO3a deficiency increases the sensitivity of CML stem cells to TKI therapy in vivo. Thus, FOXO proteins play critical roles in the resistance of CML stem cells to TKI therapy [53, 52].
One molecule downstream of FOXO that is critical for the maintenance of CML stem cells is the transcription factor B cell lymphoma 6 (BCL6) [54, 55]. BCL6, which has a BTB domain, was originally identified as a proto-oncogene in diffuse large B cell lymphoma. Homodimerization of the BCL6 BTB domain forms a lateral groove motif, which is required to bind to the silencing mediator for retinoid and thyroid hormone receptor (SMRT) and N-CoR corepressors, thereby controlling expression of target genes [56]. BCL6 contributes to the maturation of mature B cells that is mediated by transcriptional repression of p53 and survival of pre-B cells. In addition, in BCR–ABL-transformed pre-B cell acute lymphoblastic leukemia (Ph+ ALL), BCL6 is induced by TKI treatment, preventing cell death by suppressing p53 [54]. In human CML, the upregulation of BCL6 by TKI is restricted to CD34+ cells. BCL6 deficiency sensitizes CML cell lines to TKI therapy in vitro [55]. Pharmacological inhibition of BCL6 transcription activity by a recombinant peptide leads to cell cycle arrest and apoptosis in CML stem cells. Thus, the FOXO–BCL6 axis contributes to the maintenance of CML stem cells.
The promyelocytic leukemia protein (PML), an essential component of PML bodies [57], is also involved in cell cycle regulation of CML stem cells [58]. PML is highly expressed in immature CML cells, whereas its expression is barely detected in differentiated neutrophils in CML patients. PML-deficient CML stem cells undergo the cell cycle in vivo and in vitro, associated with the impairment of CML stem cell function. Although it has been reported that PML suppresses mTOR activity via the inhibition of the Rheb–mTOR association [59], the defective phenotypes of CML stem cells are mediated by mTOR activation. Pharmacological PML downregulation by arsenic forces CML stem cells to enter the cell cycle, enhancing their sensitivity to chemotherapy.
2.4 Metabolism
HSCs utilize glycolysis rather than mitochondrial oxidative phosphorylation, whereas oxidative phosphorylation becomes dominant in hematopoietic progenitors; enhanced glycolysis results in the generation of a low level of ATP energy that is associated with low ROS [7]. Dysregulation of mitochondrial activity and ROS generation in HSCs causes loss of stem cell function. The glycolytic metabolism in HSCs may be supported by a mechanism of aerobic glycolysis similar to that observed in cancer. One of the key factors for aerobic glycolysis is a protein complex containing hypoxia-inducible factor (HIF) 1, which plays a critical role in the cellular metabolic response to hypoxia [60]. HIF1-α, whose protein stability is regulated by oxygen-dependent prolyl hydroxylases, forms a protein complex with a stable partner, HIF1-β. In hypoxic conditions, the upregulation of HIF1-α enhances glycolytic flow by inducing several key enzymes that enhance glycolysis. HIF1-α also inactivates pyruvate dehydrogenase kinases, which inhibit the conversion of pyruvate to acetyl-CoA, resulting in the suppression of the influx of glycolytic metabolites into the mitochondrial TCA cycle. Several studies using genetically engineered mouse models have indicated that the Meis1–HIF1 axis is essential for glycolytic metabolism in HSCs and therefore for maintaining HSC function [61–63], although there are controversies [64, 65]. The functions of the HIF2 complex, which consists of HIF2-α and HIF1-β, appear to overlap with those of HIF1, because HIF2-α is also induced by hypoxia. Gene knockdown experiments in human HSCs showed that HIF2-α, but not HIF1-α, is essential for HSC maintenance [66]. Thus, although the precise roles of these HIF complexes in HSC maintenance in vivo are complicated, appropriate regulation of metabolic conditions appears to be important for HSC maintenance. In a mouse CML model, HIF1-α is dispensable for the generation of CML-like disease [67]. However, when the leukemia cells lacking HIF1-α are transplanted into recipient mice, the development of CML in the mice is inhibited, indicating that HIF1 is essential for the maintenance of CML stem cells. Comparison of phenotypes of HIF1-α deficiency between CML and normal hematopoiesis shows that CML stem cell maintenance depends more on HIF1-α than does the maintenance of normal HSCs. These data may be consistent with the fact that the expression of HIF1-α is induced by BCR–ABL1. Thus, HIF1 may be a therapeutic target for CML therapy.
The regulation of lipid metabolism is also important for the maintenance of CML stem cells. The plasma concentration of leukotriene B4 (LTB4), a dihydroxy fatty acid derived from arachidonic acid through the 5-lipoxygenase (5-LO) pathway [68, 69], is increased in a mouse CML model [70]. In CML stem cells, Alox5, an enzyme that synthesizes LTB4 in the 5-LO pathway, is upregulated. Interestingly, the upregulation of Alox5 in CML stem cells is dependent on BCR–ABL, but not on its enzymatic activity. Experiments with Alox5-deficient mice show that Alox5 is essential for the maintenance of CML stem cells, whereas it is dispensable for that of normal HSCs. The pharmacological inhibition of Alox5 by a selective 5-LO inhibitor, zileuton, also reduces CML stem cell number and prolongs survival of CML mice. Another molecule active in lipid metabolism, stearoyl-CoA desaturase-1 (SCD-1), is also involved in CML stem cell maintenance [71]. SCD-1, a key enzyme in fatty acid metabolism, is downregulated in CML stem cells, and its deficiency causes progression of CML disease. Because SCD-1 expression is induced by peroxisome proliferator-activated receptor γ (PPARγ), PPARγ agonists induce SCD-1, resulting in the suppression of CML stem cells.
2.5 Microenvironments
The presence of BCR–ABL-expressing cells changes the cytokine and chemokine levels in the BM microenvironment. Cytokines, including IL-1α, IL-1β, IL-6, G-CSF, and TNF-α, and chemokines, including MIP-1α, MIP-1β, and MIP-2, are upregulated in the BM in CML mouse models, and the CXCL12 level is reduced [72]. Some of these altered cytokines and chemokines have an impact on the fate or behavior of stem/progenitor cells in normal hematopoiesis and CML.
The level of IL-6, a proinflammatory cytokine, is elevated in the serum of CML patients [73, 74]. Myeloid-lineage cells in CML secrete IL-6, and the secreted IL-6 acts directly on both leukemic and normal multipotent progenitor cells (MPPs) by a paracrine loop. This biases the lymphoid differentiation of MPPs toward myeloid differentiation, resulting in the expansion of myeloid CML cells. The production of IL-6 by myeloid CML cells is controlled by BCR–ABL, because inhibition of BCR–ABL activity with TKI treatment downregulates the IL-6 expression level. In a study using IL-6 knockout mice, the decrease of IL-6 expression level delayed CML disease onset and restored the lymphoid differentiation, which showed aberrant pro-B cell features.
CXC motif chemokine ligand 12 (CXCL12/stromal cell-derived factor-1 [SDF-1]) and its receptor CXCR4 are important for the maintenance and retention of normal HSCs in BM [75]. During embryogenesis, CXCL12/CXCR4 signaling is essential for homing of HSCs and progenitor cells to BM and for the production of B-lymphoid cells [76, 77]. In adult hematopoiesis, CXCL12/CXCR4 signaling is essential for the maintenance and retention of HSCs in BM and for the production of B-lymphoid cells [78–81]. Whereas CXCR4 is expressed in several hematopoietic cell types, CXCL12 is expressed in stromal cells, osteoblasts, endothelial cells, nestin-expressing cells, and CXCL12 abundant reticular (CAR) cells. Studies of CXCL12 deletion in these CXCL12-expressing cells have demonstrated that CAR cells and endothelial cells function as a niche component. A study of conditional CXCL12 knockout mice showed that CXCL12 regulates the cell cycle status and number of HSCs in BM. Furthermore, treatment of mice with G-CSF reduces the CXCL12 level in BM. This downregulation of CXCL12 decreases HSC retention in BM and induces the mobilization of HSCs into peripheral blood [82]. In BM of both CML model mice and CML patients, increased production of G-CSF by CML cells downregulates CXCL12 [72]. This decrease of CXCL12 level results in the reduced homing of CML stem cells to BM and reduced retention there, leading to enhanced mobilization of CML stem cells into peripheral tissues. Another study reported that imatinib treatment upregulates the CXCR4 expression level in CML cells, suggesting that this signal may contribute to the resistance of CML stem cells to TKI therapy [83]. Thus, although the pathophysiological effects of the CXCR4/CXCL12 pathways on CML stem cells are still unknown, microenvironmental factors mediated by this signal critically affect CML stem cell behavior.
Recently, it was reported that the α-chain of the IL-2 receptor, also known as CD25, is expressed on some CML LICs in a CML mouse model [84]. Furthermore, expression of CD25 was also confirmed in the CML stem cell fraction, but not in the CML progenitor fraction, in patients. Importantly, CD25 is not expressed in healthy HSC and progenitor cells in mouse or human. In human CML patients, CD25 expression is elevated in the accelerated phase and in the blast crisis phase compared with the chronic phase, suggesting that CD25 signaling may contribute to CML progression. Studies using a mouse CML model showed that CML stem cells (FcεRIα−lineage marker−Sca-1+c-Kit+ cells; F−LSK) consisted of two populations of cells, CD25 positive and CD25 negative. Although these cells can interconvert, CD25+F−LSK cells actively proliferate and have higher leukemia initiating capacity than CD25−F−LSK cells. Furthermore, CD25+F−LSK cells secrete higher levels of IL-4, IL-6, IL-13, and TGF-β, supporting the maintenance of CML disease. Although the serum IL-2 level is not elevated in CML mice, CML CD25+F−Lin− cells colocalize with IL-2+Lin+ cells in BM, suggesting that mature cells may function as niche cells for CML stem cells with the support of the IL-2/CD25 pathway. Administration of IL-2 to CML mice accelerates their death, and human IL-2 increases the colony-forming capacity of human CML samples. In contrast, genetic ablation of IL2ra or administration of a monoclonal antibody against CD25 or IL-2 increases the survival of CML mice. Thus, the IL-2/CD25 axis contributes to the maintenance of CML stem cells and progression. Moreover, the combination of TKI and anti-CD25 antibody treatments reduces the numbers of CML cells, including CML stem cells, in BM and spleen. Therefore, the IL-2/CD25 axis is expected to be a direct therapeutic target for the eradication of CML stem cells.
TGF-β signals are involved in the quiescence of HSCs [48, 85]. Glial cells play a critical role in the activation of latent TGF-β, supporting the quiescent status of HSCs in BM. Although HSCs lose their stem cell properties when FOXO proteins are inactivated during in vitro culture with cytokines, TGF-β can keep HSCs quiescent without a loss of biologic potential; this is associated with the nuclear localization of FOXO3a, indicating that TGF-β may regulate HSC function through FOXO activity. CML stem cells also exhibit activated Smad-2/Smad-3 in vivo [52]. Treatment with a TGF-β inhibitor in vivo induces relocation of FOXO3a into the cytoplasm and sensitizes CML stem cells to TKI therapy in CML-bearing mice. However, another study reported that the overexpression of TGF-β observed in transgenic mice that express the receptor for parathyroid hormone in osteoblastic cells also results in the suppression of CML stem cells [86]. These data suggest that the roles of TGF-β are complicated and that fine-tuning of the TGF-β signal may be important for supporting CML stem cell maintenance. Although it is unclear whether TGF-β affects CML stem cells directly or indirectly, manipulation of this signal may contribute to the development of a therapeutic approach for CML patients.
2.6 Epigenetic Modification
Multistep process of cell differentiation is controlled by epigenetic mechanisms [13]. Epigenetics is a regulatory mechanism by which gene expression is increased or decreased by DNA methylation or posttranslational modification of histone core proteins. Histone modifications of acetylation, methylation, and phosphorylation are involved in changes in the ability of regulatory transcription machinery proteins to access the chromatin of genomic DNA, thereby controlling gene expression. Histone deacetylation is involved in the maintenance of CML stem cells, and histone deacetylase (HDAC) inhibitors have been developed and are recognized as promising medications for CML disease. Treatment of CML stem cells with HDAC inhibitors increases the acetylation levels of H3 and H4 [87]. Treatment of CML stem cells with HDAC inhibitors or imatinib decreases phosphorylation of BCR–ABL, and the combination further suppresses the phosphorylation, leading to apoptosis. Interestingly, the combination induces more apoptosis in CML stem cells than in normal HSC and progenitor cells. HDAC inhibitors efficiently reduce the number of CML stem cells in vivo in mouse CML models or mouse models using xenografts of human CML cells. Gene expression profile analyses show that HDAC inhibitor treatment reduces sets of genes regulating the undifferentiated state (e.g., the Hox–Myc and Wnt-related pathways), cell cycle regulation, protein translation, and the cellular stress response. E2F, Ying Yang 1, and NRF1/2 are reduced, whereas G protein-coupled receptors are increased. Thus, modifications of histone acetylation are critical for the expression of a variety of genes that are involved in CML stem cell maintenance.
SIRT1 is a mammalian homologue of yeast silent information regulator 2 (Sir2), and both are NAD-dependent histone deacetylases. SIRT1 functions as a histone deacetylase of histones H4K16 and H1K26 and thus regulates chromatin modification. However, it also deacetylates numerous nonhistone proteins that are involved in transcription, cell cycle, and DNA repair. SIRT1 is upregulated by BCR–ABL via the activation of STAT5; therefore, SIRT1 expression in CML stem cells is higher than in HSCs [88, 89]. Consistent with this, SIRT1 plays critical roles in the generation and maintenance of CML stem cells, although the precise role of SIRT1 in normal HSCs is controversial. In a mouse model in which the BCR–ABL gene is introduced into hematopoietic cells, SIRT1 deficiency suppresses CML. Whereas mice receiving BCR–ABL-transformed wild-type BM cells develop CML disease within 3–4 weeks, disease development is significantly delayed in mice receiving cells derived from SIRT1-deficient BM cells. In human CML stem cells, knockdown of SIRT1 induces apoptosis and inhibits proliferation. Pharmacological inhibition of SIRT1 enhances sensitivity of CML stem cells to imatinib-induced apoptosis, but it does not remarkably affect normal HSC/progenitor cells. SIRT1 inhibition enhances acetylation of p53, which induces its transcriptional activity, leading to fewer CML stem cells. Interestingly, SIRT1 is also involved in the acquisition of genetic mutations in the BCR–ABL gene induced by imatinib treatment [90]. When KCL22 cells, a human CML cell line, were treated with imatinib, the cells initially died from apoptosis, followed by regrowth after 2 weeks in vitro subsequent to the acquisition of a BCR–ABL mutation. However, inhibition of SIRT1 inhibited the imatinib resistance. In contrast, inhibitors of class I and II HDAC, such as trichostatin A, did not show such inhibitory effects on imatinib resistance. SIRT1 inhibitors, but not class I/II HDAC inhibitors, also suppress γH2AX focus formation induced by DNA damage-inducing reagents. Furthermore, DNA damage repair pathways are affected by SIRT1 through the acetylation of Ku70, which is involved in nonhomologous end joining, and NBS1, which mediates homologous recombination, generating gene mutations. Thus, although it is unknown whether histone modification by SIRT1 is involved in regulating CML stem cell behavior, this molecule is essential for the maintenance of CML stem cells.
2.7 Kinase Activity-Independent BCR–ABL Signals
CML stem cells are able to survive TKI treatment, despite expressing higher levels of BCR–ABL than other subpopulations [24]. For example, when the TKI inhibition of BCR–ABL activity was evaluated by analyzing the inhibition of Crk-like protein (CrkL) phosphorylation, CrkL phosphorylation was inhibited in CML stem cells and progenitors to a similar degree, and CML stem cells treated with imatinib showed cell growth and survival comparable to those of HSCs; however, imatinib had remarkable effects on cell cycle arrest and apoptosis in CML progenitors [91]. These data indicate that CML stem cells are insensitive to TKI. One of the explanations is the presence of signaling pathways that are activated by BCR–ABL in a manner independent of its kinase activity. Although, as described above, Alox5 is upregulated by BCR–ABL, this upregulation is not prevented by TKI treatment, indicating that BCR–ABL controls CML stem cells via Alox5 independently of its kinase activity [70]. BCR–ABL also suppresses protein phosphatase 2A (PP2A), which activates β-catenin independently of its kinase activity [92]. The phosphatase activity of PPA2 in CML stem cells is lower than that in normal HSCs, due to higher expression of an endogenous PP2A inhibitor, SET. A chemical compound, FTY720, inhibits the association of SET with the catalytic subunit of PP2A, causing the activation of PP2A. The activation of PP2A by FTY720 or PP2A overexpression induces apoptosis in CML stem cells, whereas it does not affect normal HSCs, because the effect requires BCR–ABL. Therefore, PP2A-activating drugs are expected to be effective for targeting TKI-refractory CML stem cells.
2.8 Conclusion
Accumulating evidence indicates that CML stem cells are maintained by multiple signals. Some of the signals are involved in normal HSCs, indicating that CML stem cells and HSCs have similar properties. Therefore, we believe that increased knowledge of stem cell biology will accelerate progress in leukemia research. At the same time, detailed analyses have revealed the presence of molecules or signals that contribute to the maintenance of CML stem cells but not HSCs, providing an ideal therapeutic window. Future studies on CML stem cells are sure to give rise to new concepts in CML therapy.
Related posts:
 Biomarkers for Determining the Prognosis of CML
Biomarkers for Determining the Prognosis of CML
 Recommendations for the Management of CML in the Era of Second-Generation TKIs
Recommendations for the Management of CML in the Era of Second-Generation TKIs
 Goals of CML Treatment in the Tyrosine Kinase Inhibitor Era
Goals of CML Treatment in the Tyrosine Kinase Inhibitor Era
 Updated European LeukemiaNet Recommendations for the Management of CML
Updated European LeukemiaNet Recommendations for the Management of CML
 The Role of New TKIs and Combinations with Interferon-α for the Treatment of CML
The Role of New TKIs and Combinations with Interferon-α for the Treatment of CML
 Roles for Signaling Molecules in the Growth and Survival of CML Cells
Roles for Signaling Molecules in the Growth and Survival of CML Cells
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


