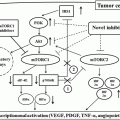
Fig. 3.1
Molecular characterization of clear-cell renal cell carcinoma PBRM1
3.2.2 Mutations of Chromatin Enzymes
The importance of chromatin dynamics and its dysregulation in tumorigenesis is increasingly appreciated since the discovery of frequent mutations in chromatin regulators, including in RCC. Earlier studies of hereditary and sporadic ccRCC have pointed to the existence of ccRCC-related genes other than VHL [63, 101]. Furthermore, in vitro studies using mouse embryo fibroblast cells or human tubular cells have shown that VHL inactivation alone paradoxically induces senescence [123] and VHL +/− mice fail to form RCC in mice model [27], whereas tubule-specific inactivation of VHL only shows cystic degeneration instead of renal tumorigenesis [115]. All these suggest that loss of VHL function alone is not sufficient to cause renal tumorigenesis. Recent high-throughput profiling has identified recurrent mutations including chromatin modifiers such as polybromo 1 (PBRM1) [112], SET domain containing 2 (SETD2 or KMT3A), lysine (K)-specific demethylase 5C (KDM5C or JARID1C), lysine (K)-specific methyltransferase 2D (KMT2D or MLL2) [17], and AT-rich interactive domain-containing protein 1A (ARID1A) [112] and also BRCA1-associated protein-1 (BAP1) in ubiquitin-mediated proteolysis pathway [31, 76]. Subsequent whole-exome and whole-genome studies further confirm these findings ([88], 15). Several key chromatin regulators involved are highlighted and further discussed below.
3.2.2.1 PBRM1 and SWI/SNF Complex
PBRM1 is the second most frequent mutated gene in ccRCC, ranging from 32 to 41 % [15, 88, 112]. The vast majority of PBRM1 mutations are indels or nonsense, typical of tumor suppressors including VHL. Furthermore, a family with four cases of ccRCC has recently been shown to be associated with germline PBRM1 mutation and loss of wild-type copy of chromosome 3p in the tumors [6]. PBRM1 encodes the BAF180 protein, an accessory subunit of the SWI/SNF (PBAF) chromatin remodeling complex [85] which is implicated in replication, transcription, DNA repair, and control of proliferation and differentiation [85]. The SWI/SNF subunits can be grouped into three categories of proteins: (1) enzymatic, (2) core, and (3) accessory. Distinct SWI/SNF complexes, consisting of PBAF and BAF variants, are formed by combinatorial assembly of a central ATPase, the core subunits, and selected accessory proteins. The latter dictates the specificity of the SWI/SNF complex function [85, 118], and BAF180 falls into the accessory group of the PBAF variant. Interestingly, the PBRM1 counterpart in the BAF variant, ARID1A which encodes BAF250a, has been previously linked to ovarian clear-cell carcinoma, mutated in 50 % of the tumors with similar pattern like PBRM1 in ccRCC [43, 117]. The fact that both are clear-cell tumors, which reflects the high glycogen and lipid contents in their cytoplasm, may point to a common biological link between the functions of these two SWI/SNF complexes. In ccRCC, although the mutations of ARID1A are significantly lower (around 2–4 %), 60–70 % of ccRCC tumors exhibit significantly lower ARID1A mRNA and protein expression according to one study [58], suggesting that decreased expression of BAF250a may contribute significantly to ccRCC tumorigenesis. Interestingly, one study has shown association of PBRM1 mutations with significant increase in ccRCC recurrence [16] and tumor-related death, while the other association of decreased BAF250a expression with higher tumor stage and grade [58]. Obviously further studies are warranted to address the role(s) of SWI/SNF complexes in ccRCC tumorigenesis.
3.2.2.2 BAP1 and Ubiquitin-Mediated Proteolysis Pathway
By whole-exome sequencing, Guo et al. identify several mutated genes from the ubiquitin-mediated proteolysis pathway (UMPP), a pathway that includes VHL along with 135 other genes [31]. A positive correlation found between the alterations in the UMPP and overexpression of HIF1α and HIF2α in ccRCC tumors suggests that alterations in the ubiquitin-mediated proteolysis pathway may contribute to ccRCC via the α-subunits of HIF [31]. Besides VHL, notable among the mutated genes in the UMPP is BAP1, which is located in the short arm of chromosome 3. Its mutations are mainly nonsense, frameshift, or splicing [76] accompanied by the loss of heterozygosity in ccRCC. Its germline mutations have also been associated with hereditary ccRCC predisposition. [80]. Originally discovered as an interaction partner of BRCA1, it regulates DNA damage response, and loss of BAP1 sensitizes RCC cell lines to γ-irradiation treatment [76]. Similar to PBRM1, BAP1 also plays a role in chromatin biology. It binds to transcription factor host cell factor 1 (HCF1) via UCH37-like domain. This interaction may sustain the formation of complexes between histone modifiers and transcription factors and regulate cell-cycle progression [67, 109]. Besides that, it can also form part of polycomb-repressive deubiquitinase complex (PR-DUB) by interacting with ASXL1. This complex deubiquitylates H2A and represses HOX gene expression [89]. Importantly, unlike the germline mutations found in melanoma and mesothelioma of which most mutants have intact UCH domain, the BAP1 mutations in ccRCC are evenly disrupted throughout the whole open reading frame (ORF). Studies have shown that its loss correlates with higher tumor grade associated with mammalian target of rapamycin (mTOR) activation [44, 76]. This correlation may have potential predictive value for prognosis and drug response.
Interestingly, another tumor suppressor gene that is inactively mutated in ccRCC and located in the frequently deleted chromosome 3p is SETD2, which encodes a histone H3 lysine-36 methyltransferase associated with methylation of active chromatin. Mutations found in either SET2 or JARID1C, a chromatin modifier, lead to decreased expression of the respective gene compared to normal kidney tissue. Furthermore, multiregional genetic analysis reveals that SETD2 and JARID1C harbors multiple distinct and spatially separated inactivating mutations within a single tumor, supporting their potential tumor suppressor roles in ccRCC [24]. SETD2 mutations found in RCC result in changes to chromatin accessibility and RNA processing defects [96].
Indeed, the relationship between the four chromosome 3p genes, VHL, SETD2, BAP1, and PBRM1, in ccRCC tumorigenesis is intriguing. First, BAP1 and PBRM1 tend to be mutually exclusive and anticorrelate in ccRCC. Using both a local database and publically available database, Kapur et al. showed overall survival to be significantly shorter for patients with BAP1-mutant tumors than for patients with PBRM1-mutant tumors [44]. A small subset of patients who have both PBRM1– and BAP1-mutant tumors appear to have worse overall survival. Interestingly, Wang et al. show that even though knockout of BAP1 fails to develop tumor just like the VHL inactivation models, combined inactivation of the VHL and BAP1 leads to RCC development [115]. These results imply that additional mutations are needed to drive ccRCC development in VHL-mutant cases, a concept that has been proposed by several lines of studies. Obviously, further functional studies to parse the biological relevance of the four genes and their interactive roles in ccRCC tumorigenesis are scientifically interesting, and tumors harboring different mutations or combination of mutation may have distinct clinical phenotypes and therapeutic implications.
3.2.2.3 Mutations of Other Chromatin Modifiers
Besides the four recurrently mutated genes in chromosome 3p, there are several other chromatin modifier genes which are mutated at a frequency of 1–4 % of ccRCC [15, 17, 110]. These include the histone methylases (i.e., MLL2) and the histone demethylases (i.e., JARID1C, JARID1D, and UTX). The size of MLL genes is large and it is possible that mutations in these genes are “passenger” mutations that arise due to random background mutation frequency. However, arguing against the random selection hypothesis is that the family of MLL genes is frequently mutated in many tumor subtypes [7, 87, 124]. In addition, JARID1C regulates histone H3 lysine 4 trimethylation (H3K4Me3) levels of HIF target genes in VHL-mutant ccRCC cells. JARID1C expression is also regulated by HIF, and depletion of JARID1C in these cells significantly promotes tumor growth, suggesting that these mutations contribute to ccRCC tumorigenesis by dysregulation of HIF-related pathway [71]. Overall, the discovery of frequent chromatin modifier mutations in ccRCC presents ample of opportunities for further research in the field of tumorigenesis and hopefully will lead to another level of understanding of these cancers.
3.2.3 Intra-tumor Heterogeneity
The traditional view of cancer development is that of clonal expansions of cells that acquire selective advantage through multiple genetic alterations acquired during its development [75]. Subsequently many studies have demonstrated that accumulation of mutations leads to divergent development of subclonal cancer cells in same tumor [26, 29, 65, 122]. Gerlinger et al. recently establish intra-tumor heterogeneity in ccRCC by multiregion exome sequencing in primary metastatic disease [24, 25]. In the study, nonsynonymous nucleotide alterations considered as potential driver mutations and found in at least one region are used to construct phylogenic trees divided into trunk, internal branches, and terminal branches. Gene inactivation of VHL is present in each case and mapped onto the trunks of phylogenetic trees. Mutations of PBRM1 are located on the trunks, suggesting that inactivation of PBRM1 also occurs at an early stage of tumor development. Overall the other driver mutations are located on the branches of the phylogenetic trees. These results demonstrate a branched rather than linear evolution in ccRCCs, similar to the branched evolution described in other types of cancers [3, 5, 8, 70, 98, 102]. An important implication of these results is that in designing drug treatment strategies, targeting mutations mapped on the trunk of phylogenetic tree may provide more preferable results than those targeting subclonal driver events [122].
The results of Gerlinger et al. also show that when multiple regions are considered for each case, the prevalence of most driver mutations is higher than single case as a whole. For instance, TP53 gene mutations are found in 2–6 % of single cases or biopsies, but up to 40 % of cases harbor the mutations when multiple regions are assessed. These data suggest that a single biopsy is unlikely to represent the full set of mutations present in a particular cancer leading to underestimation of their associated alterations. Furthermore, the relevance of subclonal driver mutations may contribute to failure of therapies [23]. Overall the evidence surrounding intra-tumor heterogeneity demonstrates the complexity and challenges in implementing precision oncology. Further understanding of the underlying biology and mechanisms of tumor heterogeneity may provide insights and help guide appropriate therapeutic strategies.
3.3 Molecular Characterization of Papillary RCC
Papillary RCC constitutes about 10–15 % of all RCC cases. It is further divided into two subtypes, type 1 and type 2, based on histological criteria [2, 19, 42]. Type 1 tumors have small cuboid cells that are arranged in a single or double layer. Type 2 tumors consist of large eosinophilic cells that are arranged in an irregular or pseudostratified manner. Type 1 pRCCs are relatively indolent and are associated with patient survival rates of approximately 90 %. In contrast, upward of 50 % of individuals with type 2 pRCC succumb to the disease within 10 years [30, 47, 78, 114]. Although the histological classification requires expert evaluation, the classification is supported by cytogenetic, gene expression, and mutational profiles that exist between these two subtypes [20, 30, 47, 61, 120].
3.3.1 Type 1 Papillary RCC (Fig. 3.2)
3.3.1.1 Copy Number Alterations
Type 1 pRCC is characterized by frequent gains of chromosomes 3q 7, 12, 16, 17, and 20 [20, 30] demonstrated by cytogenetic studies and gene expression-based deduction of chromosome changes. These findings are further confirmed by cluster analysis of SNP array data which shows nearly universal gain of chromosomes 7 and 17 and less frequent gain of chromosomes 2, 3, 12, 16, and 20 [61].

Fig. 3.2
Molecular characterization of type 1 papillary renal cell carcinoma
3.3.1.2 MET Pathway
The molecular genetics of type 1 pRCC is based on studies of hereditary pRCC that is associated with germline mutations of MET tyrosine kinase receptor, and somatic mutations in MET are also observed in up to 20 % of sporadic type 1 pRCC [61, 90, 91]. However, amplification of chromosome 7, which contains the locus of MET, and overexpression of MET are found in most of sporadic type 1 papillary tumors [1, 61]. The majority of the MET mutations are in the tyrosine kinase domain, but recently an alternate MET RNA transcript lacking its canonical exons 1 and 2, which may result in ligand-independent MET activation, is also found in some of the cases [61]. Besides the amplification of the MET locus, a member of the leucine-rich repeat kinase family, leucine-rich repeat kinase 2 (LRRK2) located in chromosome 12, is also frequently amplified and overexpressed in type 1 papillary tumors [62]. Mutations of LRRK2 are well characterized as a cause of autosomal dominant Parkinson’s disease, whereas upregulation of LRRK2 is observed in inflammatory diseases such as leprosy [125] and Crohn’s disease [4]. MET and LRRK2 cooperate during tumor growth via the mTOR and STAT3 pathway to promote cell growth and survival, and ablation of LRRK2 reduces downstream MET signaling in pRCC [62]. The central role of MET in type 1 pRCC indicates the targeted use of MET inhibitor such as foretinib, a multikinase inhibitor that targets VEGFR-2 and MET, as well as other receptors. Clinical trial of advanced stage hereditary and sporadic pRCC has seen an improvement in disease-stabilization rate and progression-free survival in patients treated with foretinib with minimal toxicity [12].
3.3.2 Type 2 Papillary RCC (Fig. 3.3)
3.3.2.1 Copy Number Alterations
Compared with type 1 pRCC, type 2 tumors harbor variable chromosome abnormalities but at different frequency. It has less gains of chromosomes 7, 12, and 17p but more frequent losses of chromosomes 8p and 9p associated with poorer survival [30, 61].

Fig. 3.3
Molecular characterization of type 2 papillary renal cell carcinoma
3.3.2.2 FH and NRF Pathways
Besides chromosome abnormalities described above, it often contains additional ones of no specific pattern, and this cytogenetic complexity may be a reflection of the more aggressive nature of this cancer type. Again, much of our understanding of type 2 pRCC comes from studies of hereditary leiomyomatosis and renal cell cancer (HLRCC) syndrome, which is caused by germline mutations of fumarate hydratase (FH), a member of the Krebs cycle. Individuals afflicted with HLRCC develop type 2 pRCC, uterine fibroids, and cutaneous leiomyomatosis (fibroid skin tumors) at high frequencies [56, 84, 104, 107]. These tumors were found to favor the Warburg-like metabolic shift to glycolysis-dependent metabolism and increased expression of hypoxia-related genes [105, 121]. Inactivation of FH leads to accumulation of fumarate that can compete against 2-oxyglutarate and inhibit PHD-mediated hydroxylation of HIFα proteins [40]. This results in stabilization of HIFα as in the case of ccRCC, but when examining their gene expression pattern, it is very obvious that the molecular signature of type 2 pRCC is rather different from that of ccRCC: the latter is predominantly of angiogenesis and metabolism while the former NRF2 pathway [72]. This leads to the demonstration that excessive fumarate, due to the inactivation of FH, is translocated into the cytosol where it reacts with cysteines of KEAP1 altering its conformation and subsequently releasing NRF1 and NRF2 from the cytoskeleton. Free NRF1 and NRF2 can then be translocated to the nucleus, where they can bind to antioxidant response element (ARE) and drive the expansion of genes such as AKR1B10 and NQO1 [72]. This pathway is further confirmed by pathway analysis of both microRNA and mRNA signatures, which clearly identifies the NRF2 pathway as a distinguishing feature of type 2 tumors [61]. Indeed, high expression of NRF2-regulated or ARE-controlled genes can be used as biomarkers with KRIB10 as a useful diagnostic biomarker [72] and NQO1, a prognostic biomarker signifying worse prognosis [61]. As inactivating mutations are uncommon in sporadic cases, it is hypothesized that members of the NRF2 pathways may be involved which is borne out by the discovery of mutations in members of the NRF2 pathways including NFE2L2, CUL3, KEAP1, and SIRT1 [51, 61, 73].
3.3.2.3 CDKN2A Alterations
High-throughput profiling has identified CDKN2A alterations in mainly type 2 pRCC (focal loss of 9p21, mutations or hypermethylation). As expected from its function, CDKN2A alterations lead to both increased levels of phosphorylated Rb and increased expression of cell-cycle-related genes. The overall survival in the patients with CDKN2A altered tumors is significantly shorter than those without CDKN2A alterations [61].
3.3.2.4 TFE3 and TFEB Fusion Genes
In a subset of type 2 pRCC, recent TCGA network has identified gene fusions involving TFE3 or TFEB, which are known to be associated with pRCC in young patients [45]. But the mean age in this TCGA study is 54 years suggesting that these fusions should be taken into account in any type 2 pRCCs [61]. In all cases with these fusions, increased mRNA expressions of known TFE3 or TFEB transcriptional targets such as CTSK, BIRC7, DIAPH1, and HIF1α are confirmed suggesting that these fusions are probably driver alterations that contribute to their tumorigenesis.
3.3.2.5 DNA Methylations and Mutations of Chromatin Enzymes
Epigenetic aberrations have been identified as important contributors of human carcinogenesis. One of them is global genome hypermethylation, resulting in suppression of tumor suppressor genes, described as CIMP [108]. DNA methylation analysis has identified CIMP, including hypermethylation of the CDKN2A promoter, in a subset of tumors with decreased FH mRNA expression. The tumors are predominantly type 2 pRCC and harbor either germline or somatic mutation of FH. These CIMP-associated tumors are associated with worst survival and increased expression of glycolysis-related, pentose phosphate pathway-related, fatty-acid synthesis-related genes [61].
Just like in ccRCC, several multiple recurrently mutated genes involved in the chromatin remodeling process have been identified in type 2 pRCC. These include SMARCB1, PBRM1, and ARID1A in the SWI/SNF complex and SETD2, KDM6A, and BAP1 in chromatin modifier pathways [51, 61]. Unlike the ccRCC, only a portion of cases with PBRM1, SETD2, and BAP1 mutations show loss of chromosome 3p where all three genes are located. The mutual exclusivity of PBRM1 and BAP1 mutations and the frequent co-occurrence of PBRM1 and SETD2 point to the intricacy and complexity of their roles in this tumor type. Further investigation is warranted to understand how mutations of these chromatin regulators impact cancer-specific gene expression in this tumor type.
3.4 Molecular Characterization of Chromophobe RCC (Fig. 3.4)
Chromophobe RCC (chRCC) comprises approximately 5 % of all renal cancers. The most distinctive and defining feature of this subtype is a perinuclear clearing (i.e., halo). The tumor cells typically show a relatively transparent cytoplasm with a fine reticular pattern – this chromophobic feature therefore gives rise to its name as chRCC [53, 69]. However, about 30 % of chRCC show eosinophilic pattern [53, 55, 103] with mitochondrial accumulation. Prognosis in localized chRCC is better than in clear-cell or papillary RCC, but metastatic disease still carries a poor prognosis without any obvious targeted therapy [46, 86]. It forms part of Birt-Hogg-Dube (BHD) syndrome characterized by cutaneous fibrofolliculomas, pulmonary cysts, and sometimes papillary RCC [106]. It is associated with germline mutations of folliculin gene (FLCN) in chromosome 17p11.2, and several reports suggested that this gene plays a tumor suppressor role in a number of cellular pathways, including PGC1α-related mitochondrial biogenesis [33, 48], TFE3/TFEB transcriptional regulation [35, 36, 77], and TGF-β signaling pathway [35, 36]. We have recently generated a mouse model with conditional knockout of proximal tubule-specific Flcn, and the mice develop multiple types of kidney tumors including chRCC which are associated with activation of mTOR and TGF-β signaling pathway [11]. Furthermore, inhibition of mTOR pathway with rapamycin can suppress the tumor growth in these knockout mice suggesting that AKT-mTOR signaling pathway plays an important role in tumorigenesis of BHD syndrome and may potentially be targeted therapeutically [11, 32, 119].

Fig. 3.4
Molecular characterization of chromophobe renal cell carcinoma
3.4.1 Copy Number Alterations
Several studies have previously reported multiple chromosomal aberrations, including the loss of whole chromosomes 1, 2, 6, 10, 13, 17, and 21 [39, 79]. Consistent with those data, SNP array analysis conducted by TCGA network shows that in 66 sporadic primary chRCC, the vast majority of tumors have loss of at least one copy of chromosomes 1, 2, 6, 10, 13, and 17 [18]. They also find losses of chromosomes 3, 5, 8, 9, 11, 18, and 21 at lower but significant frequencies. Because there are relatively few somatic mutations identified in chRCC, whether some of these altered chromosomal regions harboring key cancer genes may contribute to its tumorigenesis remains to be investigated.
3.4.2 Genomic and Mitochondrial Alterations
Interestingly, no mutations of FLCN gene are found in sporadic chRCC, and only TP53 (32 %) and PTEN (9 %) were identified as significantly mutated genes [18]. Besides PTEN, other members of the MTOR pathway that are mutated, at a frequency of <5 %, include MTOR, TSC1, and TSC2 pointing to the involvement of this pathway. Previously, frequent mutations of TP53 and mitochondrial genome have been reported in chRCC [21], and its benign counterparts, oncocytoma, are known mitochondriopathies, which are diseases related to abnormal function or mutations of mitochondria [52, 93]. Indeed, mutations of mitochondria DNA are identified in complex I of the electron transport chain (18 % of cases), especially in MT-ND5 (10 %) [18], and chRCCs harboring these mutations are linked to feature of mitochondria accumulation in cytosol and eosinophilic histology [18] similar to oncocytoma. Since these mutations are associated with loss of complex I activity [22, 66], the mitochondria accumulation has been thought as results of compensatory mechanisms for inefficient oxidative phosphorylation [97]. However, the expression levels of genes in Krebs cycle and electron transport chain are upregulated in tumors harboring these mutations [18, 22, 93]. Furthermore, the expression of PPARGC1A which encodes a regulator of mitochondrial biogenesis, PGC1α, is also increased [18]. These results suggest the existence of a metabolic shift, and these cancer cells generate much of ATP through increased mitochondria biogenesis to support the tumor growth [18].
3.4.3 Kataegis and TERT Promoter Alterations
Whole-genome sequencing conducted by TCGA network reveals ketaegis pattern, a phenomenon of localized hypermutation, in a subset of chRCC [18]. From the comparison between chRCC with and without a strong ketaegis pattern, they find very high expression of TERT which encodes the catalytic subunit of telomerase whose role is to maintain telomere ends by addition of the telomere repeat TTAGGG. Previously, deregulation of telomerase has been identified in a wide variety of cancers with upregulation of the enzyme TERT [34] which appear to play an important role in tumor progression. In these cancers, increased TERT activity is associated with point mutations [37, 38], gene amplification [13, 116], and germline polymorphisms [82]. Interestingly, in chRCC, it is the structural rearrangements involving the TERT promoter region that are associated with its strong upregulation [18], even though the underlying mechanism of how this upregulation occurs remains to be elucidated. The spectrum of events involved in the structural rearrangements includes tandem duplication, inversion, deletion, and interchromosomal translocation.
Related posts:
 Predictive and Prognostic Markers in Metastatic Renal Cell Carcinoma
Mammalian Targets of Rapamycin Inhibitors: Temsirolimus and Everolimus
Predictive and Prognostic Markers in Metastatic Renal Cell Carcinoma
Mammalian Targets of Rapamycin Inhibitors: Temsirolimus and Everolimus
 Forthcoming Drugs for Metastatic Renal Cell Carcinoma Therapy
Forthcoming Drugs for Metastatic Renal Cell Carcinoma Therapy
 Optimization of Therapy by Pharmacokinetic–Pharmacodynamic Analyses
Optimization of Therapy by Pharmacokinetic–Pharmacodynamic Analyses
 Treatment Overview
Treatment Overview
 Hereditary Renal Cell Carcinoma
Hereditary Renal Cell Carcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


