Fig. 4.1
(a) This infiltrating astrocytoma has hyperchromatic elongated astrocytic nuclei as often seen in the prototypic infiltrating astrocytomas but a significant proportion of tumor cells exhibit abundant globose eosinophilic cytoplasm (‘gemistocytic morphology’). (b) The IDH-1 R132H-specific immunostain is strongly positive with diffuse cytoplasmic immunoreactivity. (c) A significant proportion of tumor nuclei are positive for p53 immunostain. (d) Loss of nuclear immunoreactivity is observed with ATRX immunostain (arrow shows internal positive control in endothelial cells)
Together with Death-domain associated protein (DAXX; chromosome 6p21.3), ATRX is a core mediator of a chromatin remodeling complex necessary for the incorporation of histone variant H3.3 into the telomeres of chromosomes. Telomere maintenance is required for chromosomal integrity in the setting of numerous cell divisions associated with long-term tumor growth. ATRX/DAXX complex-mediated chromosomal maintenance has been implicated in telomere stability and its alteration results in alternative lengthening of telomeres (ALT), a telomerase-independent pathway for telomere maintenance that has been recognized in a significant subgroup of malignancies. Mutations in DAXX or ATRX impair the heterochromatic state of the telomeres, probably because of reduced incorporation of chromatin onto H3.3 histones. TP53 mutations play a complimentary role with genomic instability and ALT, since tumor cells presumably then have the capacity to evade apoptosis and become immortalized [38, 41, 44–49]. Nearly all ATRX-mutated cases of diffuse glioma also harbor TP53 mutations and it is thought that TP53 mutations occur first and predispose toward the acquisition of ATRX alterations [38]. Others have shown that the ALT phenotype in astrocytomas is correlated with a younger patient age; loss of ATRX expression by immunohistochemistry; p53 immunoreactivity; IDH mutations; and absence of epidermal growth factor receptor (EGFR) amplifications [50].
Prognostic markers for IDH-mutant astrocytomas will need to be better defined in order to stratify risk for this population and there is potential that additional genetic events may provide additional value. ATRX may be one such marker, since IDH-mutant, 1p/19q-intact WHO grade II gliomas with ATRX loss have been shown to have a longer median progression free survival (PFS; 4.4 years), and median overall survival (OS; 12.7 years) compared to IDH-mutant, 1p/19q-intact, ATRX-wt subgroup (PFS, 2.2 years and OS, 6.9 years), consistent with previous survival analyses [27, 51, 52]. A subset of IDH-mutant, 1p/19q-intact infiltrating gliomas have focal gains of 4q12 (platelet-derived growth factor receptor alpha; PDGFRA), 12q14 (CDK4), or 8q24 (MYC), providing additional markers for future investigation [16, 53].
4.4 The Molecular Signature of Oligodendrogliomas
Oligodendroglioma is the archetypal brain tumor with a molecular signature. While past studies primarily based on histomorphologic classifications emphasized the correlation of 1p/19q co-deletion with the oligodendroglioma phenotype and its chemosensitivity, more recent studies have stressed that the combination of IDH mutation and 1p/19q co-deletion is definitional rather than just an association [9, 16, 27, 42]. Thus, while TP53 mutations and ATRX alterations characterize IDH-mutant astrocytomas, oligodendrogliomas are defined by IDH mutations and 1p/19q co-deletions, but not ATRX and TP53 alterations, highlighting the relatively strict molecular dichotomy of IDH-mutant diffuse gliomas [16, 27, 42]. Therefore, assessment of the chromosomal arms 1p and 19q, in conjunction with TP53, ATRX and IDH mutations is essential in the diagnostic algorithm that effectively stratifies diffuse gliomas [27, 42, 51] (Figs. 4.2 and 4.3). Among the diffuse gliomas, IDH-mutant, 1p/19q co-deleted oligodendrogliomas have the longest median PFS (5.6 years) and OS (15.3 years), a finding supported by many [24, 27, 52, 54].
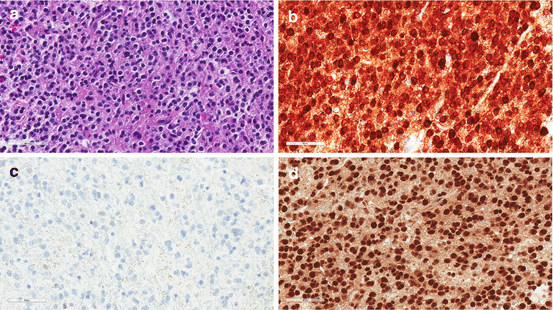

Fig. 4.2
(a) This low grade oligodendroglioma is comprised of tumor cells with round monomorphous nuclei and perinuclear cytoplasmic clearing (‘halos’). (b) The IDH-1 R132H-specific immunostain is strongly positive with diffuse cytoplasmic immunoreactivity. (c) The p53 immunostain is negative. (d) ATRX immunostain highlights intact nuclear expression
Fig. 4.3
(a) General view (Karyoview) of an IDH-mutant infiltrating glioma arising in the left frontal lobe of middle age male showing whole arm losses of 1p and 19q, and consistent with Oligodendroglioma, WHO grade II. This array detected the IDH1 R132H mutation (green dot in chromosome 2). (b and c) show the detailed view of chromosomes 1 and 19, respectively, and their corresponding whole-arm losses of 1p and 19q
Reifenberger et al. first reported that oligodendrogliomas showed a high frequency of loss of heterozygosity on the short arm of chromosome 1 (1p) and the long arm of chromosome 19 (19q) [55]. Subsequent studies demonstrated that the signature 1p/19q co-deletion is mediated through an unbalanced translocation t(1:19)(q10:p10) followed by the loss of the derivative chromosome, resulting in whole arm deletions of 1p and 19q [9, 56, 57]. Fluorescent in situ hybridization (FISH) for 1p/19q became a popular method for assessing co-deletions and is still widely used. However, it is important to realize that FISH only recognizes focal losses specific to the probes [42, 58]. Since focal losses can occur on 1p and 19q without whole arm losses, particularly in the setting of genomic instability in high grade gliomas, tests that assess only focal losses will occasionally lead to false positive results, potentially leading to the inappropriate diagnosis of oligodendroglioma [59]. Clinical tests that evaluate the entire arms, such as cytogenomic microarrays, reduce this possibility [42].
Although the R132H IDH1 mutation is the most frequent IDH mutation in oligodendrogliomas, there is a slightly higher frequency of IDH2 mutations than in IDH-mutant astrocytomas. Other molecular-genetic alterations that occur in IDH -mutant, 1p/19q co-deleted oligodendrogliomas are inactivating mutations of the tumor suppressor genes far-upstream binding protein 1 (FUBP1) gene and in the human homolog of the Drosophila capicua (CIC), on chromosomes 1p31.1 and 19q13.2, respectively. FUBP1 encodes a DNA binding protein involved in c-Myc regulation and CIC is a downstream component of the receptor tyrosine (RTK) pathway. Mutations in FUBP1 and CIC occur secondary to the unbalanced translocation with a frequency of 20–30% and 46–83%, respectively. These mutations are exceedingly rare in astrocytomas and are mutually exclusive with TP53 and ATRX mutations [9, 16, 49, 60–63]. At present, the prognostic or predictive significance of FUBP1 and CIC mutations in oligodendrogliomas remains unclear although a recent study found that outcomes of 1p/19q co-deleted gliomas were not altered by these mutations [62].
A well-known histopathologic mimic of anaplastic oligodendroglioma is the small cell variant of GBM, which needs to be distinguished since they have such differing clinical features. Small cell GBMs harbor EGFR (chromosome 7q12) amplifications in about 70% of the cases [9, 58, 64–66], whereas these amplifications are not seen in oligodendrogliomas and are mutually exclusive with IDH mutations and co-deletions of 1p/19q [59, 62]. Furthermore, imbalances of chromosome 7 and losses of chromosome 10q23 (phosphatase and tensing homolog; PTEN) in the context of a high grade glioma supports the diagnosis of GBM.
Nearly all IDH-mutated, 1p/19q co-deleted tumors also carry highly specific mutations in the TERT gene promoter (C228T or C250T), upstream of the TERT ATG start site [67–74]. TERT-p mutations are rare in diffuse gliomas with ATRX and TP53 mutations [37, 62, 67, 74]. In distinction to ALT, activating mutations in the TERT-p result in enhanced telomerase activity and lengthening of telomeres. While several reports describe concordance as high as 100%, in the TCGA study of lower grade gliomas, 96% of IDH-mutant, 1p/19q co-deleted tumors carried TERT-p mutations, while only 4% of IDH-mutant, 1p/19q intact tumors showed this mutation [16]. However, TERT-p mutations are also common in up to nearly 90% IDH-wt GBMs [67–73]. Nearly all IDH-wt infiltrating gliomas with chromosome 7 gain and chromosome 10 loss harbor TERT-p mutations or exhibit upregulated TERT expression [37]. TERT-p mutations carry an unfavorable prognosis in the absence of IDH mutations (IDH-wt GBMs) and a favorable prognosis in the presence of IDH mutation and 1p/19q co-deletion (oligodendrogliomas). Although ATRX and TERT-p mutations are nearly mutually exclusive, rare cases have been reported with both or neither [67]. Among TERT-p mutated gliomas, there is no difference in telomere length between IDH-mutant and IDH-wt cases. However, telomeres are longer in ATRX altered gliomas than those with TERT-p mutations [37].
Thus, the molecular landscape of oligodendroglioma includes mutations in IDH and TERT-p in conjunction with whole arm losses of chromosomes 1p and 19q. Gliomas harboring these three mutations have classic oligodendroglioma phenotype and have prolonged OS [67, 75]. Other genes mutated in this subset include NOTCH1, PIK3CA, PIK3R1, ZBTB20, and ARID1A. Inactivating mutations in NOTCH1 are only rarely identified in IDH-mutant, 1p/19q intact or IDH-wt infiltrating astrocytomas [16, 38, 61]. Other than a 1p/19q co-deletion, very few recurring whole arm copy number alterations (CNA) have been identified in oligodendrogliomas [16].
4.5 Molecular Signatures Argue Against Mixed Lineage Gliomas
The “mixed gliomas”, including oligoastrocytoma and GBM with oligodendroglioma component (GBM-O), have historically suffered from considerable interobserver variability in classification and grading. The 2007 WHO Classification recognized mixed gliomas as oligoastrocytomas grades II-III, as well as GBM-O, WHO grade IV, and defined them as diffusely infiltrating gliomas composed of two distinct neoplastic cells [1]. Nevertheless, in recent years numerous investigations have concluded that mixed gliomas can be usually classified as either astrocytomas or oligodendrogliomas at the molecular-genetic level and have questioned the need for the diagnosis of oligoastrocytoma [2, 16, 26, 27, 30, 35–42, 51, 58, 60, 61, 65, 67–71, 75–78]. While IDH-mutant gliomas are characterized by co-deletions of 1p/19q and TERT-p mutations or by TP53 and ATRX mutations, there is no current molecular signature for oligoastrocytoma [2, 27, 38, 42]. In the TCGA analysis, the majority of tumors diagnosed as oligoastrocytomas were IDH-mutant and had TP53 mutations (IDH-mutant astrocytomas); the remainders were found to be IDH-mutant and 1p/19q co-deleted (oligodendroglioma) or IDH-wt [16]. Others have found that most oligoastrocytomas had molecular features of oligodendrogliomas [42]. Similarly, genomic and transcriptomic studies of GBM-O have concluded that they represent either anaplastic oligodendrogliomas, IDH-mutant GBMs or IDH-wt GBMs at the molecular level, casting doubt on the need for a GBM-O designation [3]. Only rarely are cases encountered that exhibit a genuine composite of distinct tumor types, made of discrete areas of oligodendroglioma and astrocytoma, each harboring their hallmark genetic makeup [79]. It is fully expected that the application of molecular tests will result in decreased interobserver and interinstitutional variability in the diagnosis of diffuse gliomas, as well as reduced confusion related to the clinical management that has been associated with a diagnosis of mixed gliomas. At present, oligoastrocytomas are still recognized as a histological diagnosis in the revised 4th edition of the WHO Classification but its use is discouraged and, if used, should be followed by a not otherwise specified category (NOS) classifier to highlight that molecular testing was not performed or its results were inconclusive [4].
4.6 Molecular Signatures Identify Clinically Aggressive IDH-wt Infiltrating Gliomas
The presence or absence of IDH mutations stratifies adult infiltrating gliomas into two distinct subsets characterized by dissimilar genetic alterations and clinical behaviors, suggesting biologically distinct diseases despite histomorphologic similarities. The majority of primary (or de novo) GBMs are IDH-wt infiltrating gliomas (95%). This is in stark contrast to the grade II and III infiltrating gliomas, which are IDH-wt in only 20–25% of cases [14–16, 42]. By the currently employed histomorphologic criteria for grading infiltrating gliomas, IDH-wt grade II and III gliomas lack necrosis and microvascular proliferation, and therefore fall short of the histologic definition of GBM, yet their molecular-genetic profiles are strikingly similar to those of IDH-wt GBMs and they also display aggressive clinical behavior [16, 25]. In the TCGA analysis, grade II–III IDH-wt infiltrating gliomas had a genetic profile similar to primary (IDH-wt) GBM and exhibited a median OS of 1.7 years [16].
Given the clinical and genomic similarities, these lower grade IDH-wt astrocytomas could represent undersampled or incipient GBMs that have not yet developed the microvascular proliferation or necrosis required to be histologically diagnosed as a WHO Grade IV tumor [16, 38, 42, 80, 81]. In a recent study of 160 IDH-wt grade II and III astrocytomas, Reuss et al. found that 78% were molecular equivalents to conventional IDH-wt GBM, with similar frequencies in TERT-p mutations, 7p gain/10q loss, amplifications of EGFR or combined 10q/13q/14q co-deletion. A median survival of 19.4 months was noted, consistent with the TCGA analysis. Furthermore, if those grade II and III astrocytomas with H3 mutations were included, then 87% of these IDH-wt astrocytomas were molecularly and clinically indistinguishable from GBM [80]. Lower grade IDH-wt infiltrating gliomas have a much lower frequency of TP53 mutations than IDH-mutant astrocytomas. While 94% of IDH-mutant, 1p/19q intact infiltrating gliomas harbored TP53 mutations, only 25% of IDH-wt infiltrating grade II-III gliomas are TP53 mutated, similar to the frequency observed in IDH-wt GBMs [15, 34].
Other genetic alterations frequently associated with IDH-wt GBMs and lower grade gliomas include those involving PTEN, EGFR, MDM4, CDK4, NF1, PIK3CA, RB1, PTPN11, PIK3R1, and CDKN2A [16]. More recently, Di Stefano et al. reported FGFR-TACC fusions in approximately 3% of lower grade IDH-wt infiltrating astrocytomas, a frequency similar to that seen in primary GBMs, providing additional evidence of the similarity of clinical behaviors between these entities. FGFR-TACC fusions were not present in IDH-mutant gliomas and were mutually exclusive with EGFR amplifications, but often co-occurred with CDK4 amplifications [82]. Overall, as compared to the IDH-mutant counterparts, IDH-wt gliomas have greater activation of signaling through EGFR, MET, and BRAF; upregulated cell cycle activators; and reduced cell cycle inhibitors [81]. IDH-wt gliomas also have upregulation of transcription factors known as master regulators, as well as their target genes [37].
A small subset (currently estimated a less than 1%) of adult low grade infiltrating IDH-wt gliomas harbor BRAF V600E somatic mutations on chromosome 7q34 resulting from a substitution of valine by glutamic acid at codon 600 (V600E) and are thought to represent a distinct clinicopathologic entity with an improved prognosis [83–85]. BRAF V600E mutations, which constitutively activate the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) signaling pathway, are far more frequent in grade I, non-infiltrative gliomas, pediatric diffuse gliomas and epithelioid GBMs and may have therapeutic implications [32, 83, 84]. Lastly, Ceccarelli et al. described a novel subgroup of IDH-wt infiltrating gliomas that genetically and epigenetically resemble pediatric pilocytic astrocytomas and carry a favorable outcome [37]. Additional studies of IDH-wt infiltrating gliomas are necessary to address the implications of subgroups that exhibit less aggressive clinical behavior.
4.7 Primary and Secondary Glioblastomas Have Distinct Genetic Signatures
GBM is histologically defined as a high grade infiltrative astrocytoma with microvascular proliferation, necrosis or both, that has a short survival, generally less than 2 years [29, 86]. The revised 4th edition of the WHO Classification reflects the primary molecular subsets of adult GBMs by dividing them into (1) GBM, IDH-wt, (2) GBM, IDH-mutant and (3) GBM, NOS. (4). The last category is reserved for cases in which IDH assessment could not be performed or was not available. GBMs are often referred to as “primary” (or de novo) when they present to medical attention as grade IV disease as the first manifestation, and as “secondary” when they have evolved over time from a grade II or III infiltrating astrocytoma. Primary and secondary GBMs are histologically indistinguishable except for larger extents of necrosis more frequently found in the former [28]. Despite their morphologic overlap, primary and secondary GBMs differ in their genetic and epigenetic landscape, with IDH mutations being much more frequent in secondary GBMs. Secondary GBMs arise in younger patients (usually less than 45 years) and are associated with longer survivals [14, 28, 29]. Primary GBMs represent the vast majority of cases (over 90%), are nearly all IDH-wt, and are characterized by a rapid clinical onset of symptoms, most often in an elderly patient.
The genetic hallmark alterations of primary, IDH-wt GBMs include mutations of PTEN and TERT-p, gain of chromosome 7/loss of chromosome 10, deletions of CDKN2A, and amplifications of proto-oncogenes, most notably, EGFR, PDGFRA or c-MET. Although the sequence of oncogenic events in IDH-wt primary GBMs has not been determined, it has been suggested that TERT-p mutations, present in up to 90% of adult GBMs, may precede the characteristic combined gain of chromosome 7/loss of chromosome 10, seen in 60% of primary IDH-wt GBM, followed by additional oncogenic events [37, 87]. Three core signaling pathways are nearly always altered in primary, IDH-wt GBM and include (1) the receptor tyrosine kinase pathway [RTK/RAS/phosphoinositide 3-kinase (PI3K)], (2) the P53 pathway and (3) the Retinoblastoma (Rb) pathway, which are altered in 88%, 87% and 78% of GBMs, respectively [86, 88]. The most frequently altered genes in the RTK/RAS/PI3K pathway include PTEN, neurofibromin-1 (NF1), EGFR, PIK3R1, PIK3CA, and PDGFRA. Alterations in the Rb pathway include CDK4, CDK6, CCND2, CDKN2A/B and RB1. The genes frequently altered in the p53 pathway include MDM2, MDM4 and TP53 [14, 28, 29, 54, 78, 86, 88, 89]. More recently, Morris et al. reported recurrent somatic mutations in the FAT tumor suppressor (Drosophila) homolog 1 (FAT1; chromosome 4q35.2) in 20.5% of GBMs, resulting in its inactivation and leading to aberrant Wnt activation and tumorigenesis [90].
Brennan et al. has shown that 57% of GBM had evidence of mutation, rearrangement, altered splicing and/or focal amplification of EGFR, reflecting its status as a key oncogenic event in this disease [89]. Furthermore, approximately 50% of EGFR amplified tumors also harbor the variant III (EGFRvIII) deletion that leads to constitutive tyrosine kinase activation [4, 78, 91]. Approximately15–18% of primary, IDH-wt GBMs carry PDGFRA amplifications while MDM2 and CDK4 amplifications are present in 5–15% and 14–18% of the cases [4, 14, 29, 78, 89].
Deletions of the CDKN2A gene (chromosome 9p21), encoding the tumor suppressor proteins p16 (INK4a) and p14 (ARF) (activators of Rb and p53, respectively) are seen in up to 50% of GBMs [4, 14, 76–78]. TP53 and RB1 mutations and deletions are seen in 28–35% and 8–12% of primary GBMs, correspondingly [4, 14, 78, 89]. Activating mutations of PI3K are present in 12–25% of primary GBMs, with mutations in either PIK3CA or PIK3R1 driving increased enzymatic activity. Deletions or mutations in PTEN, the primary negative regulator of the PI3K/AKT signaling pathway, occur in approximately 25–35% of GBMs. Mutations or deletions of NF1, a Ras antagonist, have been identified in up to 10–18% of primary GBMs [4, 14, 29, 78, 86, 88, 89].
BRAF V600E mutations are present in less than 5% of all GBMs, but are overrepresented in epithelioid GBM with approximately 50% harboring the mutation [29]. Approximately, 5% of adult primary GBMs carry H3F3A mutations. While the frequency of BRAF and H3F3A mutations is much lower in adult gliomas than those of children, it is important to remember that both H3F3A mutations and BRAF mutations will be present in tumors that do not have IDH mutations (i.e., IDH testing will reveal an “IDH-wt” status) and testing for these mutations will need to be performed in the relevant clinical setting in order to document these distinct diseases [29, 42]. Mutation specific immunostains against the BRAF V600E (VE1) and the H3 (H3K27M) mutations are readily available and clinically useful for practical diagnostic neuropathology (Fig. 4.4).
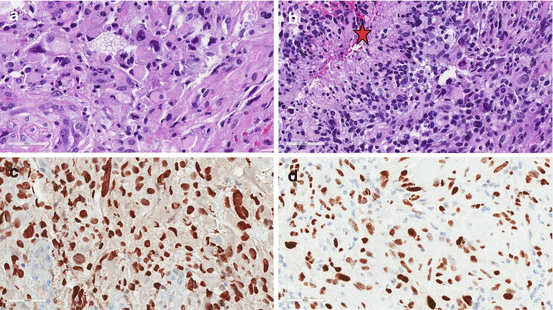
Fig. 4.4
(a and b) This GBM arose in the thalamus of a middle-aged man and was morphologically heterogeneous. The tumor was highly cellular with abundant pleomorphic cells. Pseudopalisading necrosis is evident (star in b). (c) The K27M immunostain shows strong diffuse nuclear positivity. Therefore this is best classified as a K27M-Midline GBM, WHO grade IV. (d) p53 immunostain is strongly positive as well. This GBM was IDH-wt and ATRX immunostain showed nuclear retention (not shown). TP53 and ATRX mutations often co-occur with H3K27M mutations but have the highest correlation in G34R/V-mutated GBMs
IDH mutations occur at a very low frequency in clinically diagnosed primary GBM (less than 5%) [4, 28, 92]. It is likely that IDH-mutant primary GBMs progressed from a non-symptomatic lower grade glioma that evaded diagnosis [28, 92]. As a corollary, secondary GBMs can occasionally be IDH-wt when they arise following the diagnosis of lower grade glioma; not surprisingly, secondary GBMs that lack IDH mutations usually have a poor prognosis [92]. Regardless, the IDH mutation status is more relevant to the clinical behavior of the GBM than the primary or secondary designation. Similar to the molecular-genetic makeup of their precursor lesions, IDH-mutant, secondary GBM frequently harbor TP53 and ATRX alteration: 85% of secondary GBMs are IDH-mutated and TP53 and ATRX mutations are seen in 81% and 71%, respectively [4, 40, 92]. Since these mutations occur early in gliomagenesis, additional alterations identified within IDH-mutant, secondary GBMs are likely acquired during biological progression and can serve as prognostic markers [93]. Secondary IDH-mutant GBMs contain the highest number of alternating, intrachromosomal breakpoints, consistent with chromothripsis [81]. Thus, the GBM genotypes account for biologic differences in histologically indistinguishable tumors and improves the ability to predict patient outcomes [86].
4.8 Pediatric Gliomas Are Genetically and Biologically Distinct from Their Adult Counterparts
Pediatric gliomas are most frequently either low grade and circumscribed, or high grade and diffusely infiltrative. The low grade, well circumscribed astrocytomas (most often pilocytic astrocytomas) frequently arise in the cerebellum, followed by the cerebral hemispheres, deep midline structures, optic pathway, brainstem and spinal cord [94]. Non-infiltrative or poorly infiltrative gliomas with an affinity for the temporal lobe, are also more frequent in children and include pilocytic astrocytomas (PA, WHO grade I), gangliogliomas (WHO grade I), dysembryoplastic neuroepithelial tumor (DNET, WHO grade I) and pleomorphic xanthoastrocytomas (PXA, WHO grade II or III) [4]. The histologic findings of Rosenthal fibers, eosinophilic granular bodies (EGB’s) and a low grade glioma with a biphasic appearance usually points to a diagnosis of PA and the finding of a KIAA1549:BRAF fusion is typical. This fusion event results from tandem duplications in the chromosome 7q34 region and is observed in more than 70% of PA’s, predominantly in those arising within the cerebellum, but also in other locations [95]. A temporal lobe-predominant glioma with a relatively solid growth pattern exhibiting a combination of spindle-shaped and xanthomatous cells and pleomorphic, multinucleated astrocytes in association with EGBs points to a diagnosis of PXA and the presence of a BRAF V600E mutation is supportive [96]. BRAF V600E mutations are frequent events in pediatric CNS neoplasia including gangliogliomas (20–40%), PXA’s (60–70%), DNET (30%), diffuse astrocytomas (23%) and PA’s (5–10%) [32, 97].
Most low grade neuroepithelial tumors of children have only one dominant somatic genetic event that affects protein coding. In the majority, such solitary alterations are mutually exclusive and include NF1, RAF or RAS, the receptor tyrosine kinases fibroblast growth factor receptor 1 (FGFR1; chromosome 8p11.23), and V-Myb avian myeloblastosis viral oncogene homologue (MYB; chromosome 6q23.3) or in its homologue, MYBL1 (chromosome 8q13.1) [32]. In a study of 249 pediatric low grade gliomas, which included multiple histologic entities, 90% showed recurrent somatic alterations and 83% showed rearrangements or structural alterations [98]. The most frequent genetic alterations were found in genes encoding FGFR1, the neurotrophic tyrosine receptor kinase 2 (NTRK2; chromosome 9q21.33), KRAS (chromosome 12p12.1), the receptor tyrosine kinase adaptor tyrosine-protein phosphatase non-receptor type 11 (PTPN11; chromosome 12q24), NF1 (chromosome 17q11.2), and BRAF (chromosome 7q34) [97]. Alterations of BRAF, FGFR1, PTPN11, and NTRK2 all lead to the activation of the MAPK/ERK signaling pathway, making it a primary driver of pediatric low grade gliomas [32, 99]. The most specific genotype-phenotype association was the tight linkage between angiocentric glioma and the MYB-QKI translocation [98].
Others studies have also emphasized the significance of alterations in MYB/MYBL1, FGFR1 and BRAF V600E in pediatric low grade gliomas and suggest a relationship between tumor histology and genetic alterations [32, 43, 84, 100, 101]. Qaddoumi et al. reported a high frequency of FGFR1 alterations those tumors dominated by round, regular bland “oligodendroglial-like” tumor cells, including 82% of DNETs and 40% of diffuse oligodendroglial tumors. Tumors with astrocytic differentiation and “diffuse” patterns were more frequently characterized by MYB alterations, with 41% of pediatric diffuse astrocytomas showing structural rearrangements and 87% of angiocentric gliomas showing the specific MYB-QKI fusion [100]. These findings clearly demonstrate that low grade infiltrating gliomas arising in the pediatric population are distinct from those in adults, since the IDH mutations of adult diffuse gliomas are rare in the pediatric diseases and the mutations in the pediatric diseases are not present in those of adults [43]. However, the IDH-wt status of pediatric infiltrating low grade gliomas does not imply a more biologically aggressive behavior; the rate of progression of lower grade gliomas in the pediatric population is significantly lower than their histologically comparable adult counterparts [43, 100, 102].
Among the pediatric low grade gliomas, oligodendrogliomas represent a diagnostic challenge, since they are histologically similar to adult tumors, yet do not often harbor their defining genetic alterations of IDH mutations and 1p/19q co-deletion. Only 18% of pediatric oligodendrogliomas harbor an IDH R132H mutation and only 25% exhibit 1p/19q co-deletion. Those that were IDH-mutant and 1p/19q co-deleted (‘adult-type’) occurred in older children and adolescents [102]. As described above, FGFR alterations are more frequent in pediatric oligodendrogliomas, but occur in less than half [32, 102]. BRAF alterations are absent in pediatric oligodendrogliomas, but the diffuse leptomeningeal glioneuronal tumor (known also as disseminated oligodendroglial-like leptomeningeal tumor), which was recently codified in the revised WHO Classification, are reported to harbor concurrent KIAA1549:BRAF gene fusions and 1p deletions [4, 9, 103, 104]. The precise relationship of this entity to other pediatric brain tumors, such as pilocytic astrocytoma or oligodendroglioma will require further investigation.
The high grade gliomas of childhood are also clinically and genetically distinct from those of adults. Pediatric high grade gliomas nearly always arise de novo and very rarely are the result of progression from a lower grade glioma. Although they differ from their adult counterparts in terms of location, clinical behavior, mutational landscape and gene expression profiles, they can similarly be separated into molecular subclasses [78]. Mutations targeting RTK/RAS/PI3K pathway, histone modification or chromatin remodeling and cell cycle regulation have been respectively found in 68%, 73% and 59% of these tumors, including diffuse intrinsic pontine gliomas (DIPG) and non-brainstem gliomas [105]. One of these classifications that included both adult and pediatric GBMs and used DNA methylation profiles identified six molecular classes: IDH, K27, G34, RTK I (PDGFRA), Mesenchymal and RTK II (Classic) [78, 106]. Two of these classes —the K27 and G34— were dominated by pediatric cases that harbored the respective H3F3A mutations. Korshunov et al. recently performed a large scale genomic and epigenetic integrated analysis of 202 pediatric GBMs which unexpectedly showed that 20% displayed methylation profiles similar to either low grade gliomas or PXA’s, had a better OS and were also enriched for PXA-associated molecular alterations including BRAF V600E mutations and homozygous deletions of 9p21 (CDKN2A). The remaining 162 pediatric GBMs stratified into the following four subgroups: IDH1-mutant (6%), H3.3 G34-mutant (15%), H3.3/H3.1 K27-mutant (43%), and those GBMs that were wild type for H3 and IDH (36%) [107].
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


