Lesion
Immunohistochemistry
Molecular characteristics
Nodular fasciitis
Pos: SMA, MSA, vimentin, calponin and desmin (rare)
Deletion of chromosome 2 and 13, der(15), t(2; 15)(q31; q26), tetraploidy and del (6), gains of 10p and 20q, USP6 rearrangement, t(17; 22)(p13; q13) (MYH 9—USP6 gene fusion)
Neg: CK, S100-protein, CD34, Beta-catenin
Myofibroblastoma
Pos: desmin, CD34, SMA, BCL2, CD99, CD10, ER, PR, AR, H-caldesmon (rare)
Partial monosomy of 13q and 16q monoallelic loss of FOXO1 gene, deletion of 13q14 region
Neg: CK
Desmoid-type fibromatosis
Pos: Beta-catenin (80 %, nuclear), ER (rare)
Trisomy 8 and 20, loss of 5q, 5q allelic loss, nonrandom X chromosome inactivation, Beta-catenin gene mutations, somatic or germline APC gene alterations mutation
Neg: CK, CD34
Inflammatory myofibroblastic tumor
Pos: ALK (50 %), desmin, SMA, keratin (±)
Rearrangement of the ALK gene. Common fusion partners are TPM3, TPM4, ATIC, RANBP2, CLTC, CARS, PPF1BP1, SEC31l1, RANBP2
Lipoma
S-100 protein
Coventional lipoma
Karyotypic aberrations of 12q, 6p, 13q, t(3; 12) (q27–28; q14–15) resulting in HMGA2-LPP gene fusion, 12q15 rearrangements, and t(3; 6)(q27; p21) resulting in HMGA1-LPP gene rearrangement
Hibernoma
11q13–21 rearrangements
Lipoblastoma
8q11–13 rearrangements
Chondrolipoma
t(11; 16) (q13; p12–13) resulting in C11orf95-MLK gene fusion
Spindle cell/pleomorphic lipomas
Aberration or partial monosomy of 16q, and loss of 13q14
Granular cell tumor
Pos: S-100 protein, CD68, PGP9.5, CEA and vimentin (focal)
Losses of 13q21, 4, 18q, 10, 1p, 22q
Gains of 1p32-pter, 9q33-qter, 20q, 7
Some associations with (germline PTEN mutation, PTPN11 mutation)
Neg: CK
Malignant lesions: Nonspecific complex clonal karyotype, Losses (-5, -6, -13, -15, -17, 17q-, -18 and -21)
Peripheral nerve sheath tumor
Pos: S-100 protein, GFAP, EMA (capsule), SOX10 (±), ALK (±)
Trisomy 10, loss of 9p21 (harboring INK4A/p16 and INK4A/p14 genes). 22q12 hyperploidy (EWSR1gene). May be associated with NF1 gene and NF2 gene mutations. Alterations of 9p and 22q
Neg: SMARCB1
Solitary fibrous tumor
Pos: CD34, BCL-2, vimentin, Beta-catenin, STAT6, GRIA2
Breakpoints in 12q13 resulting in NAB2-STAT6 fusion gene
Angiosarcoma
Pos: CD31, CD34, Factor VIII, MYC and prox-1 (negative in AVL), CK (focal)
Amplifications on chromosome 8q24.21 10p12.33 and 5q35.3, 8q24.21 (MYC, in secondary AS). Co-amplification of FLT4
Liposarcoma
Pos: S-100 protein
Well-differentiated LS
MDM2/CDK4
Supernumerary ring and giant marker chromosomes with 12q amplification resulting in overexpression of MDM2 and CDK4 genes
De-differentiated LS
MDM2/CDK4
Co-amplification of 1p32, 6q23 and 6q25
Myxoid/round cell LS
t(12; 16)(q13; p11) resulting in FUS-DDIT3 gene fusion. t(12; 22)(q13; q12) resulting in EWSR1-DDIT3 gene fusion
Spindle cell LS
Monosomy 7 alone, or rearrangement of 13q
Rhabdo-myosarcoma
Pos: Myogenin, MyoD1, CK, neuroendocrine markers, PAX5, ALK1, CK (±focal)
Embryonal RMS
Loss or uniparental disomy of 11p15.5, gains in chromosomes 2, 8, 11, 12, 13, and 20 resulting in gene overexpression (IGF2, H19, CDKN1C and HOTS)
Alveolar RMS
FOXO1 gene rearrangements including: t(2; 13)(q35; q14)(PAX3-FOXO1), t(1; 13)(p36; q14)(PAX7-FOXO1) and t(8; 13)(p12; q13)
FOXO1-FGFR1 gene fusion. t(x; 2)(q13; q35), t(2; 2)(q35; p23), t(2; 8)(q35; q13) resulting in PAX3-FOX04, PAX3-NCOA1 and PAX3-NCOA2 respectively. N-MYC amplification (poor prognostic feature)
Spindle cell/sclerosing RMS
8q13 rearrangements resulting in SRF-NCOA2 or TEAD1-NCOA2 gene fusions
Osteosarcoma
Pos: ALP, vimentin, SMA, desmin, S-100 protein, SATB2
Diploid to near tetraploidy, Gain in chromosome 1, loss of chromosome 9, 10, 13, and 17. Rearrangement: 1p11,1q21, 11p14, 14p11, 15p11, 17p and 19q13
Gains and losses in DNA copy numbers
Amplifications of 6p21, 8q24, and 12q14 and loss of heterozygosity of 10q21.1
TP53 and Rb genes dysfunction
Leiomyosarcoma
Pos: desmin, SMA, CK, EMA
Loss of 13q (Rb1 gene) and 10q (PTEN)
Point mutation of PTEN gene. DNA copy number gains (high-grade lesions), and
DNA copy number loss (low-grade lesions)
Nodular Fasciitis
Nodular fasciitis (NF) is a rare lesion, which classically occurs in the subcutaneous tissue of the extremities and trunk in young adults. Few cases have been reported in the breast [1, 2]. Clinically, it presents as a painful, rapidly growing, well-circumscribed lesion, which can arise within the breast parenchyma or the subcutaneous tissue. Typically, the lesion undergoes spontaneous regression within a 1–2 month period, which contributes to the low rate of cases sampled. Histologically, the lesion is composed of uniform fibroblasts or myofibroblasts arranged in short fascicles with alternating hypo- and hyper-cellular areas creating a “zonation” effect. The stroma is loose and myxoid with a “tissue culture” appearance, and contains inflammatory cells, erythrocytes and thin-walled blood vessels. Brisk mitotic activity and significant cytologic atypia may be seen, raising the suspicion for malignancy. In those problematic cases, a panel of immunohistochemical stains is helpful in making this distinction.
Immunohistochemistry
NF should always be distinguished from malignant spindle cell lesions of the breast. Negative immunoreactivity to keratins helps exclude a metaplastic carcinoma of spindle cell type (carcinosarcoma) and fibromatosis-like variant. In NF, the lesional cells are consistently positive for smooth-muscle actin with “tram track” (sub-membranous pattern of staining of myofibroblasts), vimentin, and rarely for desmin [3]. S100-protein and CD34 are typically negative.
Molecular Characteristics
This lesion was initially considered as a polyclonal reactive process. However, subsequent studies showed cytogenetic abnormalities, pointing to a clonal neoplastic process. Several case studies are noted in the literature reporting a wide array of chromosomal abnormalities in NF of the breast including, deletion of chromosome 2 and 13, derivative chromosome (15), t(2; 15)(q31; q26), tetraploidy and del (6) [4, 5] as well as gains of 10p and 20q. A more recent study of 48 cases of NF from different sites, highlighted both genomic rearrangements of the USP6 locus and a balanced translocation t(17; 22) (p13; q13) resulting in fusion of MYH 9 promoter region to the entire coding region of USP6, causing an overexpression of USP6 gene in these lesions [6]. USP6 is a proto-oncogene, located on chromosome 17p13, and is part of a large family of de-ubiquitinating enzymes, involved in processes such as intracellular trafficking, protein turnover, inflammatory signaling and cell transformation [7, 8]. Therefore, it acts as a transcription upregulator. MYH9 is a proto-oncogene, located on chromosome 22q12.3-q13, and belongs to non-muscle myosin class II family [9–12] that encodes for a protein involved in cytokinesis, cell motility, and maintenance of cell shapes and plays a role in actin network disassembly of moving cells. These genetic alterations coupled with spontaneous regression, suggest that NF may represent an interesting form of “transient neoplasia”.
Myofibroblastoma
Myofibroblastoma is a benign stromal spindle cell neoplasm composed of fibroblasts and myofibroblasts. It occurs in women between the ages of 25 and 87 years. It can also occur in men, occasionally in the setting of gynecomastia [13, 14]. Clinically, the lesion presents as a slow-growing solitary mass. It appears on imaging studies as a well-circumscribed, solid nodule that usually measures three cm or less. Calcifications are not seen. Microscopically, the lesion is well-circumscribed, unencapsulated mass with pushing borders. The classic type is composed of spindle cells that are haphazardly oriented in short fascicles within a collagenous stroma. In the epitheloid variant, the cells are larger with small nucleoli, mimicking an invasive lobular carcinoma. A prominent adipocytic component is usually present, mimicking a spindle cell lipoma. Smooth muscle, cartilaginous or osseous metaplasia may also be focally present. Normally there isn’t any entrapment of mammary ducts or lobules, which helps to differentiate it from fibromatosis.
Immunohistochemistry
The lesional cells may be positive for desmin, CD34, smooth-muscle actin, BCL2, CD99 and CD10 [14]. Negative immunoreactivity to keratins helps distinguish epitheloid myofibroblastoma from invasive lobular carcinoma, especially since both express estrogen and progesterone receptors. Androgen receptor is also positive in these lesions. H-caldesmon has been described in rare cases showing smooth-muscle differentiation [15].
Molecular Characteristics
Being a part of the same spectrum as spindle cell lipoma, myofibroblastoma shares the same cytogenetic abnormalities affecting chromosome 13, mainly partial monosomy of 13q with monoallelic loss of FOXO1 gene. Deletion of 13q14 region was also identified by fluorescence in situ hybridization [16–18]. Partial monosomy of 16q has also been reported [16].
Desmoid-Type Fibromatosis
Desmoid-type fibromatosis is a locally infiltrative lesion that arises from the fibroblasts and myofibroblasts of the breast parenchyma or, more commonly, from the pectoral fascia and extending into the breast. The lesion is more common in females and can occur at any age. It is rare and is usually associated with a history of trauma or surgery, including breast implants [19, 20]. The lesion commonly presents as a non-tender, palpable mass with occasional retraction of the overlying skin [20–22], concerning for malignancy. Histologically, the lesion resembles fibromatosis arising elsewhere in the body. It is composed of long, intersecting fascicles of bland spindle cells with infiltrating borders, and entrapment of the normal breast parenchyma. Lymphocytes are often present, especially at the tumor edges (Fig. 15.1a).
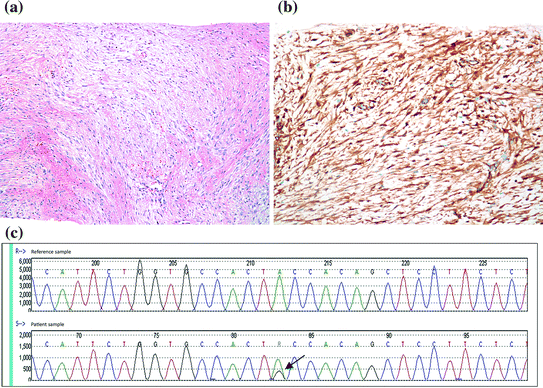
Fig. 15.1
Desmoid-type fibromatosis of the breast. a H&E stain, 100X. b Positive nuclear staining with B-catenin, 100X. c Capillary electropherogram showing a heterozygous mutation (nucleotide c.121A > AG, black arrow) involving codon 41 in exon 3 of CTNNB1 gene (encoding for beta-catenin). This results in substitution of amino acid Threonine by Alanine. Courtesy of Dr. Ediz Cosar, Department of Pathology, UMass Memorial Medical Center
Immunohistochemistry
Nuclear beta-catenin expression is a characteristic feature of these lesions (Fig. 15.1b), seen in about 80 % of cases [23]. Although this helps to differentiate mammary fibromatosis from NF, which is negative for beta-catenin, expression may be seen with other lesions including spindle cell carcinoma, phyllodes tumors, and fibrosarcoma [24, 25]. Therefore, additional markers such as keratin panels and CD34 are helpful in this differentiation as they are both negative in fibromatosis. A weak expression of estrogen receptor has also been reported in mammary fibromatosis [26].
Molecular Characteristics
Desmoid tumors have been proven to be clonal and, therefore, true neoplastic lesions rather than a reactive process. Early publications reported recurrent, nonrandom chromosomal aberrations such as trisomy 8 and trisomy 20 as well as loss of 5q [27, 28] with a suggestion of an increased risk of recurrence in cases associated with trisomy 8. Another study showed nonrandom X chromosome inactivation in some tumors, with the same inactivation patterns identified in multiple recurrent lesions, suggesting that they were derived from the same cell clone as the primary lesions. The morphologic resemblance of these tumors to deep fibromatosis elsewhere in the body is also evident at the molecular level. Recent publications [23, 24, 29] have demonstrated somatic alterations of the APC/β-catenin pathway in the majority of breast fibromatoses, similar to those seen in desmoid tumors. These included activating β-catenin gene mutations and somatic APC gene alterations, either by mutation, 5q allelic loss or both. The occasional occurrence of these tumors in patients with familial adenomatous polyposis also reflects the finding of germline mutations of the APC gene in this group of patients.
Beta-catenin, a cadherin binding protein, is encoded by a proto-oncogene, CTNNB1, located on chromosome 3 with dual function of regulating the coordination of cell–cell adhesion and gene transcriptional coactivator, as a part of the canonical Wnt signaling pathway which promotes cellular proliferation and their differentiation. The Wnt proteins bind to receptors on the cell surface called “frizzled proteins,” triggering a multi-step process to allow Beta-catenin entry into the cell. Without Wnt signaling, the Beta-catenin would be degraded because of a “destruction complex,” a composition of multiple proteins including APC and glycogen synthetase kinase-3beta (GSK-3β), a regulator of the Beta-catenin level, preventing its accumulation within the cells. The APC encoded by a tumor suppressor gene is located on long arm of chromosome 5 is a key component of this complex. Any alterations of the Wnt signaling pathway or APC gene mutation will result in an accumulation of β-catenin in the cytoplasm with its eventual entry into the nucleus to form a complex with TCF (T cell factor-1)/LEF (lymphoid enhancing factor-1) factors and thereby acting as a transcription coactivator of cellular oncogene, including cyclin D1, c-JUN and c-myc. Additionally, somatic CTNNB1 gene mutation (Fig. 15.1c) results will alter serine/threonine residues at GSK-3β phosphorylation sites. The same mutations have been identified in fibromatosis outside of the breast. An additional 21-bp deletion spanning codons 42 through 48 has also been reported [29]. All together, these mutations will prevent the destruction complex to degrade Beta-catenin within the cytoplasm, and ultimately entering the nucleus followed by gene transcription activation, which then leads to the stromal cell proliferation seen in mammary fibromatosis [23, 24, 29].
Inflammatory Myofibroblastic Tumor
Inflammatory Myofibroblastic Tumors (IMT) commonly occur in children and young adults, usually in the abdominopelvic region, lung or retroperitoneum. Only a few case reports of IMT in the breast have been published [30, 31]. Clinically, it presents as a painless, well-circumscribed mass, and histologically, the lesion is composed of fascicles of myofibroblasts with minimal cytologic atypia. A prominent mixed inflammatory infiltrate dominated by plasma cells is the hallmark of these tumors. Lymphocytes and neutrophils can also be frequent.
Immunohistochemistry
The cells in IMT stain positively with smooth-muscle actin and occasionally with desmin and/or keratin. A characteristic feature of these tumors is their immunoreactivity for anaplastic lymphoma kinase (ALK) protein. Approximately 50 % of cases stain positively for ALK [31]. Both nuclear and cytoplasmic (primarily) staining can be seen. ALK-positivity is not very specific and can be seen in other lesions such as rhabdomyosarcomas and malignant peripheral nerve sheath tumors [32, 33]. However, it helps to differentiate this inflammatory cell-rich neoplasm from mastitis.
Molecular Characteristics
The ALK gene (2p23) encodes a receptor tyrosine kinase that, under normal conditions, is only present in normal tissue. Clonal alterations involving the ALK gene have been initially described in anaplastic large cell lymphoma (ALCL). However, ALK gene alterations can also be seen in some soft tissue neoplasms. Approximately 50 % of IMT cases exhibit rearrangement of the ALK gene [34, 35]. Several fusion partners have been identified in IMTs, the most common being TPM3 at 1p23. Others include TPM4 at 19p13, ATIC at 2q35, CLTC at 17q23, CARS at 11p15, RANBP2 at 2q13, PPF1BP1 at 12p11, SEC31l1 at 4q21 and RANBP2 at 2q13. The latter may be associated with poor prognosis. All the aforementioned fusion gene rearrangements are reflected as a cytoplasmic immunostaining pattern, with the exception of RANBP2 (a nuclear pore protein) which exhibits a nuclear staining pattern. Interestingly, NMP-ALK, the most common fusion gene seen with ALCL, has not been reported in IMTs, while TPM3-ALK, ATIC-ALK, and CLTC-ALK were identified in both neoplasms, thus, representing rare examples of translocation-derived chimeric tyrosine kinases, driving both mesenchymal and lymphoid neoplasms.
Lipoma
Lipomas of the breast present as painless, solitary masses arising within the subcutaneous tissue of the skin overlying the breast rather than breast parenchyma itself [14]. Microscopically, conventional lipoma is a proliferation of mature adipocytes surrounded by a thin capsule. A conventional lipoma can be difficult to distinguish from the normal breast adipose tissue, especially in a core biopsy specimen where the capsule is not easily identifiable. Lipomas with small vessels containing fibrin thrombi are characteristic of angiolipomas, which contrary to other sites, are painless. Other variants reported in the breast include angiolipoma [36–38], spindle cell/pleomorphic lipoma [39], angiomyolipoma [40], hibernoma [41], adenolipoma [42], fibrolipoma [42], lipoblastoma [43], and chondrolipoma [44].
Immunohistochemistry
With the exception of some variants where the morphologic evidence of a lipomatous origin can be obscured, immunohistochemistry is usually of little, if any, value. The cells show diffuse positivity with S-100 protein.
Molecular Characteristics
Multiple chromosomal abnormalities have been described in lipomas. Karyotypic aberrations affecting 12q, 6p, and 13q have been described in over 80 % of conventional lipomas, while up to 87 % of spindle cell/pleomorphic lipomas have aberration of 16q. A subset of spindle cell lipomas shares the same genetic abnormality with conventional lipomas, which include loss of 13q material, particularly 13q14 [45]. This leads to a diminished expression of multiple genes, including C13orf1, DHRS12, ATP7B, ALG11 and VPS36 and C13orf1.
Multiple studies highlighted a subset of conventional lipomas with a specific t(3; 12)(q27–28; q14–15) chromosomal translocation causing a fusion of high motility group A2 (HMGA2, exons 1–3) gene and the lipoma preferred partner (LPP, exons 9–11) gene [46–51]. The high mobility group proteins are nuclear proteins that bind DNA and function as transcription cofactors, including HMGI-C (encoded by a gene on 12q15) and HMGI-Y (encoded by a gene on 6p21) [46, 52, 53]. Normally both genes are only expressed in embryos but not in adult tissue [54–57]. However, both genes are expressed in some tumors and transformed cell lines [57] including lipomas (by chromosomal segment rearrangement at either 12q15 or at 6p21) and atypical lipomatous tumors (by amplification in the ring and giant marker chromosomes at 12q15) [47, 48, 58]. Subsequent study by Kubo et al. [59] showed a reciprocal LPP-HMGA2 fusion gene in a subset of lipomas involving exons 7 and 8 of LPP gene and exon 4 of HMGA2 gene. While 12q15 rearrangements and t(3; 12) (q27–28; q13–15) seem to be the most common genetic alterations seen in conventional lipomas, in a study by Wang et al., a case of lipoma with t(3; 6)(q27; p21) causing fusion of HMGA1 to the LPP/TPRG1 intergenic region has been reported [60]. Another publication exhibited a genetic link between spindle cell lipoma, cellular angiofibroma, and myofibroblastoma, which consists of either breaks or deletions of a limited region within 13q14, in the vicinity of retinoblastoma (RB1) gene, BRCA2 and lipoma high mobility group proteins (HMGI-C) fusion partner genes (LHPF) or partial monosomy of 16q, as well as chromosomal rearrangements of 13q and 16q [16, 61]. Hibernomas have been reported to frequently harbor 11q13–21 rearrangements, which has subsequently shown to cause homozygous or hemizygous gene loss of two tumor suppressor genes MEN1 and/or AIP that present a low expression in hibernomas but high expression of genes upregulated in brown fat including PPARA, PPARG, PPARGC1A and UCP1 [62, 63].
Lipoblastomas have been reported to harbor 8q11–13 rearrangements or to exhibit excess copies of chromosome 8 which results in overexpression of PLAG1 [62, 64]. Pleomorphic adenoma gene 1 or PLAG1 is a proto-oncogene, which encodes a zinc finger protein with 2 putative nuclear localization signals. It is considered a Transcription factor whose activation results in upregulation of target genes, such as IGFII, leading to uncontrolled cell proliferation and its overexpression has been reported in pleomorphic adenoma of salivary gland as well as lipoblastomas. Chondroid lipomas have also been reported to harbor t(11; 16)(q13; p12–13) resulting in C11orf95 (chromosome 11 open reading frame 95)-MKL2 (myocardin like 2) fusion oncogene [65].
Granular Cell Tumor
Granular cell tumor is a neoplasm of the Schwann cells of the peripheral nerves. Up to 8.5 % of all granular cell tumors occur in the breast [66–68], more frequently in females and over a broad age range. Although benign, the lesion may have a worrisome radiologic appearance with irregular or speculated borders. Moreover, it can cause nipple inversion and skin retraction, further mimicking carcinoma. Grossly, the tumor has a homogenous, tan-white cut surface, and histologically, it is composed of infiltrative clusters and sheets of uniform, round to polygonal cells with indistinct cell borders and round to oval nuclei. The cytoplasm is characteristically filled with fine, periodic acid-Schiff (PAS)/diastase resistant cytoplasmic granules.
Immunohistochemistry
The lesional cells are diffusely and strongly positive for S-100 protein and CD68 [69]. Together with pan-keratin negativity, a panel of these 3 stains is helpful in differentiating this lesion from infiltrating carcinoma, especially lobular and apocrine types. There is also a strong expression of PGP9.5 and focal expression of carcinoembryonic antigen (CEA) and vimentin. The ki-67 proliferation index is typically low.
Molecular Characteristics
Comparative genomic hybridization analysis of GCT occurring within central nervous system has shown a variety of genetic changes. Overall, losses (−13q21, −4, −18q, −10, −1p, −22q) were more frequent than gains (+1p32-pter, +9q33-qter, +20q, and +7) [70]. Multiple lesions have been reported to be associated with some inherited familial syndromes such as Neurofibromatosis, Cowden syndrome (via germline PTEN mutation on chromosome 10), Bannayan-Ruvalcaba-Riley syndrome (via germline PTEN mutation on chromosome 10), Leopard syndrome (via PTPN11 mutation on chromosome 12, implicating the Ras/MAP kinase pathway) and Noonan syndrome (via PTPN11 mutation on chromosome 12, implicating the Ras/MAP kinase pathway) [71–75]. Malignant granular cell tumors (MGCT) appear to exhibit a very complex clonal karyotype. In one case report, which presents a case of MGCT of the ulnar nerve, 21 cells underwent cytogenetic analysis a very complex karyotype was reported as 44 to 47, XY, +X, del(1)(p?), del(5)(p?), add(20)(q13), +22, +mar(cp 11) [76]. In another case report of MGCT of the lateral femoral cutaneous nerve, cytogenetic analysis of seventeen cells in metaphases, showed two clonal karyotype, one complex-44 to 47, XY, +del (1) (p?), del (5) (p?), add (20) (q13), −22, +mar (cp11) and one normal-46, XY [77]. The modal number was near diploid (range, chromosome 44 to 47). In addition, occasional aberrations such as -5, -6, -13, -15, -17, 17q-, -18 and -21 were also detected. In another case report of MGCT, a karyotype of 46, XX, +X,dic (5; 15) has been reported [78]. Therefore, it appears that to this day, despite the complexity of these genetic alterations, no specific recurrent karyotype alteration has been linked to these lesions.
Peripheral Nerve Sheath Tumor
These tumors are derived from the sheath of the peripheral nerves and include Schwannoma and Neurofibroma. These tumors are almost always benign, however, rare cases of malignant peripheral nerve sheath tumor (MPNST) have been reported [79–83]. Histologically, neurofibroma is an unencapsulated lesion, characterized by a fascicular proliferation of spindle cells, with occasional mast cells and focal myxoid changes, while Schwannoma is classically an encapsulated, biphasic lesion with alternating hypercellular (Antoni A) and hypocellular (Antoni B) spindle cell areas out cytologic atypia. Cytologic atypia, mitotic activity and necrosis are features of MPNST.
Immunohistochemistry
The lesion is positive for S-100 protein and GFAP. EMA stains the capsule surrounding Schwannoma. A subset of MPNST stains positive with SOX10 and ALK, and exhibits a loss of normal SMARCB1 nuclear staining [84]. SMARCB1(INI1) is a member of SWI/SNF multi subunit chromatin remodeling complex which plays an important role in regulating transcription [85].
Molecular Characteristics
There are reports of MPNST demonstrating trisomy 10 [86, 87]. Chromosome 10 harbors many oncogenesis including PTEN, which has been implicated in numerous human malignancies. In addition, in the same study, fluorescence in situ hybridization (FISH) analysis revealed loss of locus 9p21 (encoding for proteins INK4A/p16 and INK4A/p14 which regulate cell cycle, ensuring underphosphorylation of Rb gene) in 94 % of the examined cells and 22q12 (containing cancer related gene EWSR1) hyperploidy in 83.1 % of the examined tumor cells. Alterations of 9p and 22q have also been reported with several malignancies, including MPNST [86–88]. In addition, multiple neurofibromas have been reported in association with neurofibromatosis type 1 by deletion or mutation of NF1 tumor suppressor gene located on chromosome 17q, while multiple Schwannomas are reported either in association with type 2 neurofibromatosis, by monosomy or partial loss of NF2 tumor suppressor gene on chromosome 22 or by a germline mutation of INI1/SMARCB1(located on chromosome 22) [89] in familial schwannomatosis with high risk of malignant transformation [90]. Lastly MPNST can result from 17q loss, causing an NF1 (somatic or germline) mutation [91].
Solitary Fibrous Tumor
Solitary fibrous tumor (SFT) rarely occurs in the breast, with only a few case reports [92]. This lesion presents clinically as a slow-growing mass. Histologically, it is composed of spindle cells arranged in short fascicles haphazardly arranged in a myxoid background with sometimes a prominent vascular network (hemangiopericytoma-like) creating the customary “patternless pattern” type of morphology.
Immunohistochemistry
Molecular Characteristics
A constant genetic alteration reported with this lesion includes recurrent breakpoints in 12q13 clustering near the NAB2 and STAT6 genes resulting in NAB2-STAT6 fusion gene, which will cause cell proliferation and activation of the expression of EGR-responsive genes [93, 95–97]. More recently gene expression profiling has highlighted a number of upregulated genes in SFT [96–98]. GRIA2 has been identified as one of the most common and highly expressed genes, encoding an AMPA-selective ionotropic glutamate receptor subunit which controls the membrane calcium permeability and appears to be involved in cell proliferation and motility. GRIA2 is normally involved in nervous system development, in some neurodegenerative disorders [99–101] as well as some tumors [100, 102–104]. The exact mechanism of action of GRIA2 has not been fully elucidated. A study by Ishiuchi et al. [105] suggests that GRIA2 is involved in PI3 K-independent activation of the AKT signaling pathway. While the involvement of GRIA2 in oncogenesis seems undeniable, more studies are needed to fully understand its exact mechanism of action.
Angiosarcoma and Atypical Vascular Lesions
Clinically, angiosarcomas (AS) of the breast can be divided into two subtypes. Primary angiosarcoma [106, 107] which arises de novo within the breast parenchyma. Although rare, primary AS represents the second most common mesenchymal malignancy after malignant phyllodes tumors, with an in incidence of 0.05 % of all breast malignancies [108]. Secondary angiosarcoma develops in the skin of the breast, chest wall, or the breast parenchyma, classically in breast cancer patients, who underwent axillary lymph node dissection followed by radiation therapy. The frequency of secondary AS has been notably increasing in the last two decades making it the most common radiation-associated sarcoma in the breast. This increasing number of cases reflects the increasing frequency in this treatment modality [109, 110].
Atypical vascular lesions (AVL) are vascular proliferations that develop in the skin of the breast in women who were treated with lumpectomy and radiation therapy for invasive breast carcinoma. Some believe these lesions may be the precursors to angiosarcoma, especially since a preceding or a synchronous AVL may be encountered in patients with angiosarcoma [111–119]. Clinically, AVL presents as ill-defined brown/erythematous patches or plaques on the skin of the breast within the radiation field, approximately 6 years into treatment. Microscopically, the lesion shows a dissecting, interconnecting vascular network lined by plump endothelial cells with prominent nuclei, but without significant cytologic atypia (Fig. 15.2a) [113, 118, 119]. On the other hand, AS usually presents as a bulky mass with bruise-like changes of the overlying skin. Histologically, a well-differentiated AS consists of anastomosing vascular channels lined by neoplastic endothelial cells with prominent and hyperchromatic nuclei with rare mitosis and lack of solid areas. In addition to the above findings, a poorly differentiated AS exhibits an epithelioid cell morphology, numerous mitotic figures, areas of necrosis as well as areas of solid tumor growth (Fig. 15.3).
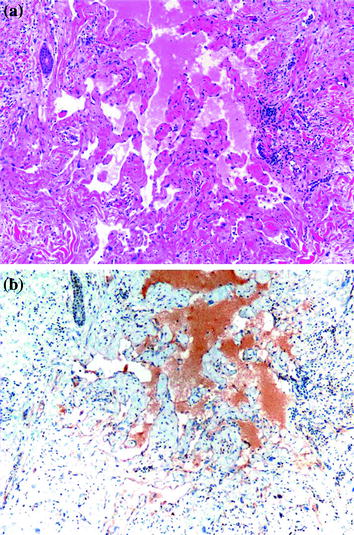
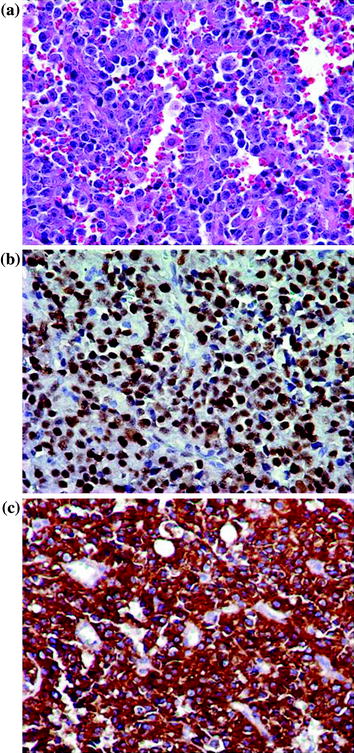
Fig. 15.2
Atypical vascular lesion of the breast. a H&E stain, 100X. b MYC stain shows negative staining, 100X. Courtesy of Dr. Kristine Cornejo, Department of Pathology, UMass Memorial Medical Center
Fig. 15.3
Post-radiation angiosarcoma of the breast. a H&E stain, 400X. b Positive nuclear staining for MYC, 400X. c Strong and diffuse cytoplasmic staining (3+) for FLT4, 400X. Courtesy of Dr. Kristine Cornejo, Department of Pathology, UMass Memorial Medical Center
Immunohistochemistry
While poorly differentiated AS is easy to recognize as malignant, a well-differentiated AS may be mistaken for pseudoangiomatous stromal hyperplasia (PASH), hemangiomas, and even with AVL. PASH is a myofibroblastic proliferation disguised in a vascular microscopic appearance. It usually presents as an incidental finding and only rarely as a mass-forming lesion. Its myofibroblastic origin can be unmasked by showing immunoreactivity to actin, desmin, calponin, progesterone receptor as well as CD34, and negativity for endothelial cell markers such as factor VIII and CD31. Hemangiomas usually present as non-palpable lesions identified on mammography. Microscopically, hemangiomas show a proliferation of variably sized, anastomosing vascular channels that infiltrate the interlobular stroma without disrupting the lobular architecture. This is an important feature differentiating it from well-differentiated AS. Differentiating AVL from a well-differentiated AS can be challenging particularly on core needle biopsies, and in samples obtained from the edge of the lesion. Immunoreactivity to MYC and prox-1 has been proposed as potential marker differentiating AVL (negative for MYC and focally positive for prox-1) from AS which is positive for both (Figs. 15.2b and 15.3b) [120]. Finally, cytokeratin may show focal expression in AS, but should not be mistaken for a spindle cell carcinoma.
Molecular Characteristics
In a study done by Antonescu et al., in which 42 samples from 39 patients with AS from diverse sites including breast (primary and secondary lesions) have been examined, these lesions were reported to exhibit an upregulation of vascular specific tyrosine kinases, including TIE1, SNRK, KDR, TEK and FLT1. In addition, 10 % of patients showed KDR mutations and these lesions were all of primary breast site (primary or secondary lesion) [121]. In another study by Manner, et al., in which samples from 28 patients with primary AS and 33 patients with secondary AS were analyzed by array comparative genomic hybridization, the authors showed recurrent genetic alterations in 22 cases, all of which presented with secondary AS. The most frequent of these alterations included high level amplifications of chromosome 8q24.21 (50 %), 10p12.33 (33 %) and 5q35.3 (11 %) [122]. These findings indicate that primary AS and secondary AS are two genetically distinct lesions, despite their clinical and histological resemblance. Few studies have also demonstrated MYC gene (8q24.21) amplification in approximately 55 % of secondary AS [123–125]. In a study comparing AS to AVL [123], MYC amplification by FISH was identified in all AS cases, in contrast to AVL where all samples were negative. Other studies showed co-amplification of FLT4 (encoding VEGFR3) which was only observed in 25 % of cases of secondary AS (Fig. 15.4) [124] and strong and diffuse nuclear staining with prox-1 which was present in secondary AS and was lacking in AVL (Fig. 15.2) [125].
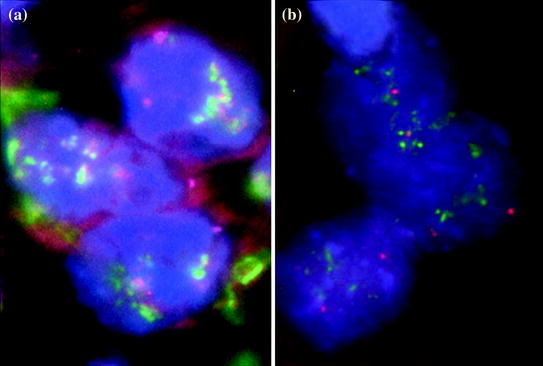
Fig. 15.4
MYC and FLT4 FISH in post-radiation angiosarcoma. a Markedly increased intensity and number of FLT4 (red) and MYC (green) signals compared to CEP8 (magenta). b Confirmation of FLT4 amplification using different probes showing increased signals of FLT4 (green) compared to CEP5 (magenta). Courtesy of Dr. Kristine Cornejo, Department of Pathology, UMass Memorial Medical Center
Liposarcoma
Liposarcomas are the most common soft tissue sarcomas; however, primary liposarcoma of the breast is an extremely rare lesion [126–127]. It commonly occurs as a heterologous component in the setting of a malignant phyllodes tumor. Rare post-radiation liposarcoma of the breast are also reported [128]. Liposarcoma usually presents as a slowly growing, sometimes painful mass. Grossly, it is a well-circumscribed mass, but may have an infiltrative edge. Of all subtypes, the well-differentiated liposarcoma/atypical lipomatous tumor is the most common tumor of the primary mammary liposarcomas. Histologically, the hallmark feature is the presence of lipoblasts, which are immature adipocytes with hyperchromatic, scalloped nuclei and cytoplasmic vacuoles.
Immunohistochemistry
Molecular Features
Atypical lipomatous tumors as well as the well- and de-differentiated liposarcomas have a supernumerary ring and giant marker chromosomes, containing amplified material from the long arm of chromosome 12, an area which harbors many proto-oncogenes leading to overexpression of MDM2 and CDK4 as well as other genes, including HMGA2, YEASTS4, CPM, and FRS2 [129–135]. Occasionally, 1q21–25 segment is co-amplified, which can cause ATF6 and DUSP12 amplification in a subset of these lesions [136]. In cases of the aggressive de-differentiated liposarcoma, in addition to the previously mentioned genetic alterations, a co-amplification of 1p32, 6q23 and 6q25 sequences can be seen, causing JUN, ASK1 and MAP3 K7IP2 amplification respectively [137, 156]. Distinct genetic alterations including t(12; 16)(q13; p11) have been reported in over 90 % of myxoid/round cell type which results in FUS-DDIT3 gene fusion, and rare cases have t(12; 22)(q13; q12) resulting in EWSR1-DDIT3 gene fusion [138]. The spindle cell type liposarcoma, has been reported to exhibit a monosomy of 7 alone or a rearrangement of 13q, without the usual 12q amplification. These data are supported by the fact that these lesions are also MDM2/CDK4 negative [139–141].
Rhabdomyosarcoma
As in other breast sarcomas, rhabdomyosarcoma occurs more commonly as focal component in older women with malignant phyllodes and metaplastic carcinomas [142, 143]. Conversely, primary rhabdomyosarcomas of the breast are very rare [144], and occur mainly in children and young adults. Although, embryonal rhabdomyosarcoma is the most common soft tissue subtype, primary breast rhabdomyosarcoma is almost often the alveolar type [144] which is histologically characterized by thin fibrous septae lined by small round blue cells in an alveolar growth pattern.
Immunohistochemistry
Alveolar rhabdomyosarcoma should be distinguished from lobular carcinoma with an alveolar growth pattern, especially that pan-cytokeratin may show focal positivity in rhabdomyosarcoma. Immunoreactivity to skeletal cell markers such as myogenin and myoD1 is characteristic [145–147]. Neuroendocrine markers [147], PAX5 [148] and ALK1 can also be positive.
Molecular Characteristics
There are no specific reports on the molecular makeup of primary breast rhabdomyosarcoma. However, the alveolar type is associated with FOXO1 gene (a member of the fork-head family, formerly known as FKHR) translocations. The most common translocation is t(2; 13)(q35; q14) resulting in PAX3-FOXO1 gene fusion. Other translocations include t(1; 13)(p36; q14), t(X; 2)(q13; q35), t(2; 2)(q35; p23), t(2; 8)(q35; q13) and t(8; 13)(p12; q13) resulting respectively in PAX7-FOXO1, PAX3-FOX04, PAX3-NCOA1, PAX3-NCOA2 and FOXO1-FGFR1 fusion gene formation [149–157]. N-MYC amplification has been proposed as a poor prognostic feature in alveolar rhabdomyosarcoma [158]. The embryonal type has been linked to either a loss or a uniparental disomy of 11p15.5 with gains in chromosomes 2, 8, 11, 12, 13, and 20, which will cause amplification of genes such as IGF2, H19, CDKN1C and HOTS [149–151]. Spindle cell/sclerosing type rhabdomyosarcoma, a recently described subtype, presents 8q13 rearrangements, which will result in SRF-NCOA2 or TEAD1-NCOA2 fusion gene formation [159].
Related posts:
 Modelling the Molecular Pathology of Breast Cancer Initiation
Molecular-Based Diagnostic, Prognostic and Predictive Tests in Breast Cancer
The Molecular Pathology of Chemoresistance During the Therapeutic Response in Breast Cancer
Molecular Pathology of Breast Cancer Metastasis
Modelling the Molecular Pathology of Breast Cancer Initiation
Molecular-Based Diagnostic, Prognostic and Predictive Tests in Breast Cancer
The Molecular Pathology of Chemoresistance During the Therapeutic Response in Breast Cancer
Molecular Pathology of Breast Cancer Metastasis
 Molecular Basis of Breast Cancer Imaging
Molecular Basis of Breast Cancer Imaging
 Triple-Negative Breast Cancer: Subtypes with Clinical Implications
Triple-Negative Breast Cancer: Subtypes with Clinical Implications
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


