3.2.2 Integrative Molecular Classification
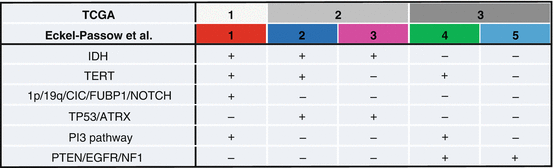
In additional efforts to construct clinically-relevant and objective tumor subclasses, two recent landmark studies have proposed classification schemes defining subgroups of lower-grade glioma (grades II and III) with distinct patterns of molecular and clinical features. Using unsupervised clustering of genomic, transcriptomic, epigenomic, and proteomic data from 293 lower-grade gliomas, an analysis from the TCGA network defined three disease subgroups according to IDH, TP53, and 1p/19q status [15]. Two groups were defined by those with IDH mutation with or without 1p/19q codeletion; the first included patients who also tended to have CIC, FUBP1, NOTCH1, PI3 kinase pathway, and TERT promoter alterations, while the second group was characterized by patients who also tended to have TP53 and ATRX mutations. The authors suggested that development of these two types of lower-grade gliomas is initiated by IDH mutation and G-CIMP establishment, then driven forward by either TP53 mutation or 1p/19q codeletion. Tumors without an IDH mutation marked a third group that was molecularly and clinically comparable to that seen in primary grade IV glioma (glioblastoma), displaying frequent PTEN, EGFR, NF1, TP53, or PIK3CA alterations.
In a second, larger study by Eckel-Passow et al. including TCGA cases as well as cases from two independent sets from the UCSF Adult Glioma Study and the Mayo Clinic, 615 lower-grade gliomas were classified into five groups according to IDH mutation, 1p/19q codeletion, and TERT promoter mutation status [16]. The classification scheme proposed by Eckel-Passow et al. is similar to that proposed by the TCGA study but incorporates TERT promoter rather than TP53 mutation status to further refine three groups into five, generating a comprehensive classification system that also incorporates higher glioma grades; a comparison of the molecular alterations that define the two classification schemes is illustrated in Fig. 3.2. The lower-grade glioma groups included tumors without any of the three alterations (7%, “triple-negative”), with IDH mutation only (45%), TERT mutation only (10%), TERT and IDH mutations (5%), and with all three alterations (29%, “triple-positive”). As in the TCGA study, subgroups were associated with characteristic acquired alterations: CIC, FUBP1, and NOTCH1 mutations were seen in triple-positive tumors, PI3 kinase pathway mutations in triple-positive and TERT mutation only tumors, TP53 and ATRX mutations in tumors with IDH mutation with and without TERT promoter mutation, and EGFR, PTEN, and NF1 mutations in triple-negative and TERT mutation only tumors. Subgroups also had characteristic patterns of copy-number changes and TCGA expression subtypes [17]; interestingly, tumors harboring IDH mutations with or without TERT and 1p/19q alterations were most closely related to the proneural expression pattern, while triple-negative or TERT mutation only tumors were more likely to display classical or mesenchymal expression patterns, indicating greater molecular similarity to primary glioblastoma.
Additionally, the five subgroups had notable differences in the distribution of histologic subtypes. These data, along with population-level incidence data, can be used to construct population-based estimates of the proportion of grade II astrocytomas and oligodendrogliomas by histology and molecular subtype [16, 18] (Fig. 3.3). According to these estimates, nearly three-quarters of grade II astrocytomas and oligodendrogliomas combined are triple-positive or have IDH mutation only. Interestingly, among grade II astrocytomas and oligodendrogliomas, triple-positive tumors tend to be predominantly oligodendrogliomas, while all other subtypes tend to be astrocytomas. It is important to note that these estimates do not include oligoastrocytomas due to the lack of available population data by grade for this histologic type, and that histological subtypes are based on traditional definitions before the 2016 updated WHO guidelines.
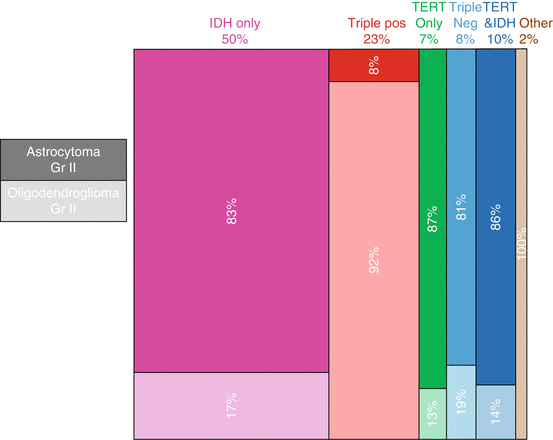
Fig. 3.3
Estimated population proportions of grade II astrocytomas and oligodendrogliomas by histology and molecular subtype. Percentages along top row denote the estimated proportions of each molecular subtype (IDH mutation only, Triple positive (IDH mutation, TERT mutation, and 1p-19q codeletion), TERT mutation only, Triple Negative (neither IDH or TERT mutation and 1p-19q not co-deleted), TERT mutation and IDH mutation, or Other) among grade II astrocytomas and oligodendrogliomas combined, and percentages within boxes denote the estimated proportions of each histological subtype within a given molecular subtype. Dark and light shading correspond to astrocytoma and oligodendroglioma, respectively. (Proportions were estimated based on data from Eckel-Passow et al. [16] and the Central Brain Tumor Registry of the United States (CBTRUS) [2] and calculated according to the method described by Rice et al. [18])
3.3 Heritable Genetic Influences
While DLGGs are most often sporadic, ~5% of glioma cases are familial in nature, and the risk of glioma development is approximately twice that in first-degree relatives of glioma patients than for the general population [19–23]. Although such familial aggregation can be a reflection of shared environmental exposures, a growing number of constitutive genetic risk factors have been identified that cause glioma or contribute to glioma susceptibility.
3.3.1 Familial Susceptibility
Rarely, gliomas are caused by single-gene hereditary cancer syndromes; for example, Li-Fraumeni syndrome can cause astrocytomas or other malignancies as a result of an inherited TP53 mutation [24]. Other glioma-causing syndromes include neurofibromatosis, Lynch syndrome, melanoma-neural system tumor syndrome, and Ollier disease [25, 26] (Table 3.1).
Table 3.1
Familial syndromes associated with increased risk of low-grade gliomaa
Syndrome | Gene(s) | Mode of inheritance | Other features/predispositions | Associated glioma subtypes |
|---|---|---|---|---|
Li-Fraumeni syndrome | TP53 | Dominant | Breast, brain cancer, soft-tissue sarcoma, other cancers | All types |
Lynch syndrome | MSH2, MLH1, MSH6, PMS2 | Dominant | Gastrointestinal, endometrial, other cancers | All types |
Melanoma-neural system tumor syndrome | CDKN2A | Dominant | Malignant melanoma | All types |
Melanoma-oligodendroglioma susceptibility syndrome | POT1 | Dominant with reduced penetrance | Malignant melanoma | Oligodendroglioma and oligoastrocytoma |
Neurofibromatosis 1 | NF1 | Dominant | Neurofibromas, schwannomas, café-au-lait macules | Astrocytoma |
Ollier disease/Maffucci syndrome | IDH1/IDH2 | Dominant with reduced penetrance | Intraosseous benign cartilaginous tumors, other cancers | All types |
In non-syndromic families, linkage studies have been used in an effort to identify glioma-associated risk loci. An early linkage analysis of a small number of individuals with varying glioma grades detected a potential familial glioma locus at 15q23-q26 [27]. A larger nonparametric linkage analysis conducted by the Gliogene Consortium in families segregating glioma of all grades found a significant linkage peak at 17q12-21.32 and suggestive peaks at 6p22.3, 12p13.33-p12.1, and 18q23 [28]; a subsequent parametric analysis supported the prior finding of a risk locus on chromosome 17q [29]. Follow-up targeted sequencing of the 17q region identified candidate missense variants implicating four genes (MYO19, KIF18B, SPAG9, and RUNDC1) of potential importance to gliomagenesis [30]. It should be noted that since the distribution of glioma grades in the Gliogene Consortium is comparable to that of non-familial cases, the majority of cases included in the aforementioned analyses had higher grade tumors [31], so whether the identified loci are specifically associated with low-grade glioma risk is unknown.
In a further effort to identify glioma-driving germline mutations, whole exome sequencing was performed in 90 members of 55 families with aggregation of glioma. The analysis identified three families, each with at least one low-grade oligodendroglioma, harboring three different mutations in protection of telomeres protein 1 (POT1) [32]. POT1 is a component of the telomere shelterin complex involved in telomere maintenance and DNA damage response, and loss of POT1 expression has been shown to cause telomere lengthening [33–35]. This study represents the first characterization of POT1 mutation as a potential glioma (particularly low-grade oligodendroglioma) predisposition syndrome, and adds to a growing literature indicating that the lengthening of telomeres is a hallmark of gliomagenesis [36].
3.3.2 Common Inherited Susceptibility Variants
A series of genome-wide association studies (GWAS) conducted since 2009 has identified common genetic variants at six genetic loci that are related specifically to the inherited risk of low-grade glioma development, and another four that appear to affect risk of all glioma grades (Table 3.2). Single nucleotide polymorphisms (SNPs) in the 8q24.21 region near long-intergenic noncoding RNA CCDC26 display the strongest associations with low-grade glioma, specifically with oligodendroglial tumor development [37–40]. Fine-mapping of the 8q24.21 region pinpointed rs55705857 as the strongest risk allele in the region, yielding strikingly large odds ratios for risk of oligodendroglial tumors as well as IDH-mutant astrocytomas (~5-fold increased risk) [41, 42]. rs55705857 maps to a highly conserved DNase hypersensitive region of CCDC26 that may be related to CCDC26 expression via transcription factor or polycomb complex binding. Interestingly, it was observed in the study by Eckel-Passow et al. that subjects carrying the risk allele at rs55705857 were at increased risk of developing gliomas in all three subgroups characterized by IDH mutation [16].
Table 3.2
Inherited variants associated with risk of low-grade glioma and all grades of glioma
Chromosome location | Candidate gene | rsID | Odds ratio | Associated tumor subtype(s) |
|---|---|---|---|---|
5p15.33 | TERT | rs2736100 | 1.35 | All glioma subtypes |
8q24.21 | CCDC26 | rs55705857 | 5.00 | Oligodendroglial, IDH-mutant astrocytic tumors |
9p21.3 | CDKN2B | rs1412829 | 1.35 | Astrocytomas grade II-IV |
10q25.2 | VTI1A | rs111696067 | 1.19 | Low-grade glioma |
11q23.2 | ZBTB16 | rs648044 | 1.25 | Low-grade glioma |
11q23.3 | PHLDB1 | rs498872 | 1.50 | IDH-mutant glioma |
12q21.2 | (Intergenic) | rs12230172 | 1.23 | Low-grade glioma |
15q24.2 | ETFA | rs1801591 | 1.36 | Low-grade glioma |
17p13.1 | TP53 | rs78378222 | 2.70 | All glioma subtypes |
20q13.33 | RTEL1 | rs6010620 | 1.40 | All glioma subtypes |
SNPs near PHLDB1 at chromosome 11q23.3 have also been associated with the risk of low-grade glioma, and specifically with the risk of IDH-mutated gliomas [39, 40, 43]. An association has been observed between inheriting a risk allele at 11q23.3 and developing gliomas in the IDH-mutation-only subclass [16]. A follow-up functional study implicated either PHLDB1 or nearby DDX6 as target genes in the locus and identified a candidate functional SNP in an enhancer element regulating DDX6 [44]. Most recently, in the largest GWAS of glioma to date, Kinnersley et al. reported the identification of four new lower-grade glioma risk loci at 10q25.2 near VTI1A, 11q23.2 near ZBTB16, 12q21.2, and 15q24.2 near ETFA [45].
Variants in four other gene regions appear to be related to the development of all grades of glioma (although some grade-specific associations have varied across studies). These include polymorphisms at 5p15.33 in TERT, 9p21.3 near CDKN2A/CDKN2B (which appear to affect risk of astrocytic tumors in particular), 17p13.1 in TP53, and at 20q13.33 near RTEL1 [16, 25, 40, 46–49]. Variants in three additional regions (3q26.2 near TERC, 7p11.2 near EGFR, and 12q23.33 near POLR3B) appear to be predominantly associated with high-grade glioma, although again reports have varied [40, 45, 46, 50, 51].
3.4 Environmental and Developmental Risk Factors for DLGG
Epidemiological studies have explored many exposures for association with glioma risk with inconsistent results, particularly for low-grade glioma development [25, 52]. Due to the rare nature of these tumors and frequent pooling of glioma grades in many previous epidemiologic studies, the evidence for associations with many factors (e.g., occupational exposures, dietary factors, anti-inflammatory agents) is limited and inconclusive, and will require larger studies with detailed exposure histories and well-annotated biospecimens for increased study validity. A summary of the established, probable, and unlikely risk factors for low-grade glioma is presented in Table 3.3. The risk factors with the best evidence for contributing to low-grade glioma risk, namely exposure to ionizing radiation and history of allergies or atopic disease, are discussed here.
Table 3.3
Non-genetic risk factors for low-grade glioma
Exposure | Direction of association |
|---|---|
Established risk factors | |
High-dose ionizing radiation | Increased risk |
Male vs. female gender | Increased risk |
White vs. African American ethnicity | Increased risk |
Probable risk factors | |
History of allergies/asthma | Decreased risk |
Probably not risk factors | |
Alcohol consumption | |
Antihistamine use | |
Cellular phone use | |
Diagnostic radiation | |
Head injury | |
Low-frequency magnetic fields | |
Smoking | |
3.4.1 Radiation Exposure
Therapeutic or high-dose ionizing radiation is a long-established cause of brain tumors in adults [53]. Although subtype- or grade-specific data are limited in published studies [25], cases of grade II glioma, specifically, among individuals previously exposed to radiation therapy have been reported [54, 55]. With regard to medical diagnostic radiation exposure, a suggestive association for an increase in low-grade glioma risk has been reported [56], but studies are limited and results inconsistent for this exposure. The impact of nonionizing radiation from cell phone use has been the subject of much research and speculation. In 2011, radiofrequency fields were classified by the International Agency for Research on Cancer (IARC) as a possible carcinogen due to the observation of increased glioma risk among heavy cell phone users [57]. Subsequent epidemiologic studies have not supported an association between cell phone use and glioma development overall [25], and studies that have stratified by glioma grade have not identified associations between cell phone use and low-grade glioma risk [58].
3.4.2 History of Allergy and Atopy
Epidemiologic studies have revealed an inverse association between allergic conditions and glioma development that is robust to an array of study designs (i.e., single site, multisite, nested case control; prospective cohort; meta-analysis) and exposure metrics [59–62]. Several recent studies have specifically assessed the association of allergy, asthma, and antihistamine use with low-grade glioma risk. Two independent studies published in 2011 found associations between allergy or asthma history and significantly reduced risks (~30–50%) of low-grade glioma development that were comparable in magnitude to associations for higher grade gliomas [63, 64]. Self-reported history of antihistamine use was not associated with low-grade glioma risk in either of these studies. In a study specific to oligodendroglial tumors, history of asthma was associated with a 50% reduction in grade II oligodendroglioma risk [65]. Other studies have reported weaker protective effects of allergy history against low-grade glioma than for high-grade glioma [66, 67].
Most recently, an analysis in the Glioma International Case-Control Study (GICC) population of the role of allergies or other atopic conditions on glioma susceptibility demonstrated a borderline significant ~20% reduction in risk in glioma associated with history of any allergy (OR = 0.79, 95% CI: 0.61–1.02). When stratified by tumor grade, the association was significant for high-grade (OR = 0.75, 95% CI: 0.58–0.98) but not low-grade (OR = 0.84, 95% CI: 0.63–1.11) glioma [62]. With regard to respiratory allergies in particular, the study demonstrated a statistically significant ~30% reduction in glioma risk (OR = 0.72, 95% CI: 0.58–0.90); stratified by tumor grade, the association was comparable for high-grade glioma (OR = 0.70, 95% CI: 0.57–0.85) but was attenuated for low-grade glioma (OR = 0.80, 95% CI: 0.62–1.03). Thus, the protective effect of allergies appears to be less pronounced for low-grade than for high-grade glioma, although it should be noted that a smaller sample was analyzed for low-grade glioma, and there is the potential for recall bias that may be differentially poor among cases, particularly among high-grade subjects. Finally, both asthma status and a history of eczema were associated with statistically significant decreased risks of glioma overall and of low-grade glioma in particular (all ~20–30% reductions), and no significant association was detected for long-term antihistamine use.
3.5 Survival
While survival after diagnosis with DLGG is better than that for high-grade glioma patients, it has not improved over the last several decades and DLGG is still ultimately a fatal disease with median survival between 7 and 12 years [3, 4]. Tumor histology and certain clinical factors have been linked to DLGG survival; however, these features are limited in their clinical value and ability to predict tumor progression and/or survival. In recent years, novel molecular biomarkers and tumor classifications have emerged that are remarkably informative for outcome prediction.
3.5.1 Histological and Clinical Prognostic Factors
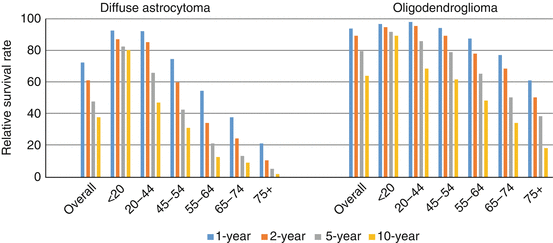
Tumor histology and several clinical variables have been shown to affect the prognosis of patients diagnosed with DLGG. Median survival is longest for patients diagnosed with oligodendroglial tumors (~12.5 years) and shortest for patients with astrocytic tumors (~7 years), with intermediate survival for oligoastrocytomas (~11 years) [3, 4]. Relative survival rates (1-, 2-, 5-, and 10-year) for diffuse astrocytoma and oligodendroglioma by age are presented in Fig. 3.4 , and clearly illustrate the poorer overall survival for astrocytoma relative to oligodendroglioma, and the worse survival among older age groups for both histological subtypes. Other prognostic variables include functional level as measured by Karnofsky performance status and tumor size and location [52]. While there is considerable observational evidence that the extent of surgical resection is associated with better survival [68], it has been noted that this may be related to differential tumor aggressiveness according to operable vs. inoperable brain locations or clinical judgment [3]. Evidence is more conclusive that early surgical resection improves survival relative to a watchful waiting approach [69, 70]. Radiation therapy has been shown to delay time to recurrence, but does not improve and may worsen overall survival [3, 71]. Data on the impact of chemotherapy with temozolomide (TMZ) on DLGG survival are limited [72–75].