Molecular Diagnostics in Hematology
![]()
BACKGROUND
The application of molecular biology and genetic techniques has greatly contributed to recent advances in hematology. Many new technologies have found utility in the clinical routine. This chapter illustrates the application of molecular techniques in the diagnosis of hematologic diseases and explains the principles and details of the most commonly used tests. The individual techniques are described in the context of specific applications; many of the methods are applied in a variety of diseases described in specific chapters of this handbook.
The most essential technologies of the next decades will include:
![]() Polymerase chain reaction (PCR) and its modification
Polymerase chain reaction (PCR) and its modification
![]() Sanger sequencing
Sanger sequencing
![]() DNA microarray technologies
DNA microarray technologies
![]() Next generation sequencing (NGS)
Next generation sequencing (NGS)
In particular, these technologies are of fundamental importance for many derived diagnostic tests, and we provide here a short overview of their general principles, while subsequent paragraphs show specific diagnostic areas and modifications. Of note is that many indirect methods of sequence variant determination, such as melting curve analysis, restriction fragment polymorphism, PCR amplification with sequence-specific primers (SSP) and hybridization with a sequence-specific oligonucleotide probe (SSOP), will be increasingly replaced by direct sequencing.
Polymerase Chain Reaction
PCR revolutionized molecular diagnostics in hematology; various modifications of this technique exist. Both DNA and RNA reverse transcribed into cDNA can be used as a template. In the presence of forward and reverse DNA primers that bind to the sequence-specific regions of the target DNA, Taq polymerase extends both strands of the DNA. Repeated cycles of annealing, extension, and denaturation lead to the exponential amplification of the targeted DNA sequence with the specificity provided by the DNA primers (Fig. 29.1). PCR primers can be designed to distinguish polymorphic sequences; primers can be labeled to facilitate detection or quantitation. Various modifications of the basic PCR technologies have been described below in conjunction with specific applications.
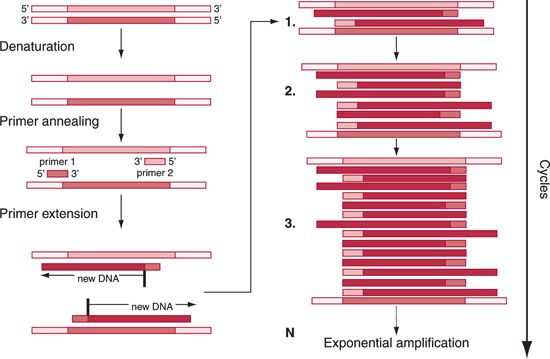
FIGURE 29.1 Principle of polymerase chain reaction.Template consists of either DNA or cDNA generated by reverse transcription of mRNA. In the presence of forward and reverse DNA primers that bind to the sequence-specific region of the target DNA,Taq polymerase extends both strands of the DNA such that repeated cycles of annealing, primer extension, and denaturation lead to amplification and accumulation of the targeted newly synthesized DNA sequence with the specificity provided by DNA primers.
Traditional Sequencing
The most popular method uses modified chain-termination method and was introduced by Sanger in the early 1970s. Sanger sequencing relies on the use of color-labeled, dideoxynucleotide chain terminators (ddNTPs). Compared to a regular reaction mixture, it consists four, regular deoxynucleotides (dNTPs) mixed with color-labeled ddNTPs. In principle, when the ddNTP is added at the end of the fragment it restricts further elongation of the DNA chain. Since the addition of the dNTPs and ddNTPs is random, a whole array of fragments of different lengths will be produced. Each fragment will be end-labeled with only one dye that represents one of four different nucleotides. Because all the fragments have different sizes, they can be separated and visualized by capillary electrophoresis. In capillary electrophoresis, each color-labeled fragment migrates according to its length, shorter fragments migrating faster. At the end of the capillary, each fragment is analyzed by a laser beam and fluorescence detector. Since each fragment reaches the detector sequentially, nucleotide sequence is reconstructed from a wavelength chromatogram; in most cases, only a small portion of the gene of interest (usually 1 exon per reaction) is sequenced. Consequently, even single-gene sequencing is extremely labor and cost intensive, requiring multiple PCR reactions and site-specific sets of primers.
New Generation Sequencing
NGS overcomes the limitation of Sanger sequencing by using a massive, parallel sequencing approach (Fig. 29.2). DNA is randomly fragmented and millions of fragmented DNA molecules are physically arrayed and attached to universal adaptors on a solid surface. A set of universal primers is then used to massively amplify and ultimately sequence millions of fragments in parallel. Elongation of each of the small fragments is similar to conventional DNA sequencing. Four different color-labeled terminator nucleotides are added sequentially, elongating the DNA chain, unincorporated nucleotides being washed off. After one terminator nucleotide is successfully added, it is able to emit one of the four distinct colors when the laser beam is applied. With the advent of ultra-sensitive charged coupled device (CCD) cameras, added nucleotides can be distinguished. Since the process is done in parallel, “reading” of each of millions of sequenced fragments is accomplished. Finally, the cycle is finished with removal of reversible terminator. By repeating this cycle multiple times (until the template fragment is fully extended), the complete sequence of the fragment, one nucleotide at the time, is completed. The lengths of the fragments are determined by the number of cycles that are needed to completely sequence a given DNA fragment. The process produces millions of short fragments (reads) that need to be organized into meaningful, organized, continuous sequences corresponding to specific target genes or chromosomal regions or transcripts (if mRNA is the starting material). Computer algorithms and sequence aligners assemble the sequences for analysis.
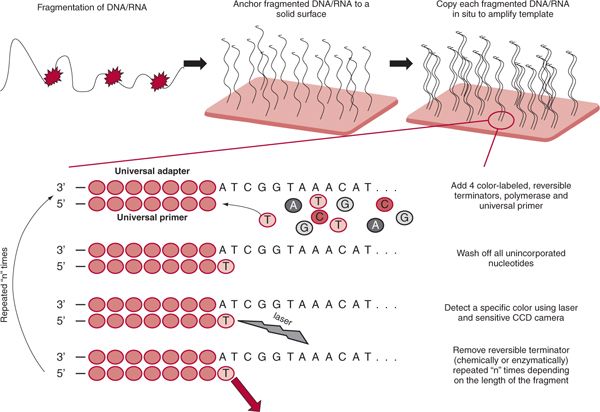
FIGURE 29.2 Principle of next generation sequencing.The first step usually consists of DNA/RNA fragmentation. It is done either enzymatically (restriction enzyme) or mechanically (sonication). Subsequently all the fragments are arrayed by hybridization to universal adapters attached to a solid surface. Successful attachment along with proper dilution guarantees only one fragment per location.This enables each fragment to be sequenced in parallel. Usually fragments are being copied prior to sequencing. Sequencing proceeds in four steps. The first step is the addition of a reversible terminator that results in elongation by one nucleotide. Secondly, all the unincorporated nucleotides are removed. Subsequently the whole reaction surface with all the fragments is scanned using the laser beam and CCD camera.The last step involves modification of the termination moiety, and the cycle can start over.The number of cycles represents the length of millions of fragments sequenced.
![]()
DETECTION OF INDIVIDUAL GERMLINE MUTATIONS/ POLYMORPHISMS
Precise diagnosis of many hematologic diseases or detection of susceptibility to develop complications depends on the identification of mutated genes. Clinically applicable methods mostly involve detection of defined mutations occurring at specific sites within the genes. Currently, most protocols use PCR to amplify the involved gene fragments. For the identification of the presence of individual mutations, various methods can be used (Fig. 29.3). For example, they are applied in routine diagnosis of genetic hematologic diseases including thalassemia and other hemoglobinopathies, hereditary familial hemochromatosis (HFE) gene mutations such as C282Y and H63D, factor V Leiden, prothrombin gene mutations G20210A, and thermolabile C677T 5,10-methylenetetrahydrofolate reductase.1,2 Similar methods can be applied for the detection of other clinically relevant mutations or polymorphisms.
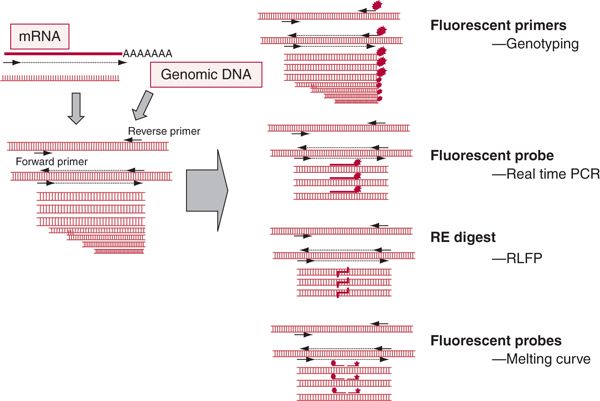
FIGURE 29.3 Application of PCR technology for the detection of a gene mutation.Various techniques based on PCR can be used for specific applications in hematology. Fluorescent primers can be used for determining the small differences in the size of the amplified product, a technique called genotyping. Fluorescent probes can also be selected to hybridize between the primer sequences of the template allowing design of real-time PCR. DNA amplicons generated in the process of PCR reactions can be used for restriction endonuclease digestion. If restriction sites for specific enzymes within the amplicon contain a mutation, restriction endonuclease digestion of the PCR product will result in fragments of different sizes, which can be resolved on either capillary or agarose gel electrophoresis. Finally, using specific fluorescent probes that hybridize to the amplified sequences, melting curves can be recorded to distinguish individual alleles.The presence of sequence differences between the probe and template results in different melting curves; these curves are recorded based on the emission of light induced by melting off the fluorescent probes from the template.
Restriction Fragment Length Polymorphism Analysis
Prior to the advent of PCR technology, traditional Southern blotting of genomic DNA followed by probe hybridization was used to detect changes in the endonuclease restriction patterns. Currently, restriction fragment length polymorphism (RFLP) analysis is used in conjunction with PCR amplification. Restriction digests can be performed either prior to or after amplification. If a mutation affects the restriction endonuclease digestion patterns, its presence can be easily demonstrated using RFLP analysis. After PCR amplification of a relevant gene fragment that carries a specific mutation, the resulting amplicons are subjected to restriction endonuclease cleavage. Using gel electrophoresis, changes in the fragment size can be demonstrated. Through comparison with a wild-type form, heterozygote and homozygote patterns can be easily distinguished. When a fluorochrome-labeled primer is used, capillary gel electrophoresis can be applied allowing for high sensitivity and throughput. Detection of HFE mutations by RFLP of PCR products serves as an example for this technique.
Melting Curve Analysis of Polymerase Chain Reaction Products
More recently, a light-cycler PCR method combined with melting curve analysis has been used for the detection of gene mutations allowing for a reduction in the workload and enabling automation. Melting curve analysis exploits the fact that even a single-nucleotide mismatch between the labeled probe and the targeted sequence significantly reduces the melting temperature. Consequently, amplicon/probe mismatches will melt off at lower temperatures different than that of matched target DNA. PCR amplification of specific gene fragments is performed in the presence of a fluorescent DNA probe or probes (anchor and sensor probe) that release light on hybridization to the internal portion of the amplicon containing the potential mutation. After completion of the reaction, the hybridized fragments are denatured; the release of the probe decreases the amount of the emitted fluorescence, a process recorded in the form of a melting curve. The shape of the melting curves identifies the presence of two normal alleles (singular curve), or heterozygotes (two peaks). For mutation homozygotes the curve is shifted, producing a singular characteristic peak. If multiple mutations are present in a gene, specific probes and primers must be applied to detect heterozygotes, homozygotes, and compound heterozygotes.
Allele-Specific Polymerase Chain Reaction
The tetra-primer amplification refractory mutation system (ARMS) PCR is one of the variants of allele-specific PCR; it allows for detection of single nucleotide polymorphisms (SNPs) as well as single-gene mutations. Two different allele-specific amplicons and a larger (non-allele-specific) control amplicon are generated by a pair of two common (outer) primers and by two allele-specific (inner) primers that have opposite orientation (allele 1–specific primer, antisense, and allele 2–specific primer, sense). Because the common primers are designed so that the mutation is located nearer one of them, the two allele-specific amplicons will have different lengths and thus easily separated by gel electrophoresis: the wild-type genotype generates two bands on gel electrophoresis, homozygous mutation generates two bands, and heterozygous mutation generates all three bands.
Direct Polymerase Chain Reaction–Amplified Product Sequencing
Alternative methods of mutation analysis include direct sequencing of PCR products. Both alleles can be easily identified, and the direct sequencing method has the advantage that it does not target a specific mutation and that all possible sequence differences can be detected within the sequenced gene region.
![]()
MOLECULAR DIAGNOSIS OF HEMOGLOBINOPATHIES
Hemoglobinopathies constitute a large group of inherited autosomal recessive hematologic disorders. While routine laboratory tests and clinical presentation are often sufficient for a proper diagnosis, molecular analysis is mandatory for the confirmation of the defect and precise characterization of the abnormal hemoglobin.3 For example, combinations of specific mutations may greatly affect the phenotype expected in the progeny. Thus, molecular diagnosis may have significant consequences for counseling affected patients, asymptomatic carriers, and prenatal diagnostics.
Traditionally, Southern blotting was used, but, recently, PCR-based methods are preferred. Allele-specific oligonucleotide (ASO) hybridization and allele-specific priming are the most commonly applied techniques. The first method relies on hybridization of ASO probes (wild type and mutant) to PCRamplified genomic DNA. In the dot-blot assay, ASO is labeled, while the reverse dot-blot technique utilizes labeled amplified DNA, allowing for simultaneous screening of multiple mutations. Allele-specific priming is based on the principle that a perfectly matched primer pair amplifies target DNA more efficiently than a mismatched pair. In the ARMS, genomic DNA is challenged with both wild-type and mutant primer sets. Multiple mutations may be simultaneously screened in a multiplex PCR assay using fluorescently labeled ARMS primers, producing products of different length that can be detected using an automated DNA analyzer. Large deletions of both the α and β globin gene may be screened using the gap-PCR, with primers complementary to the breakpoint sequences. However, for some deletion mutants, Southern blotting is still standard.
Combining all these approaches in the context of the ethnic- and region-specific distribution of globin mutations, successful molecular identification is possible in more than 90% of cases. Mutations remaining unknown after standard molecular screening may be investigated further by denaturing gradient gel electrophoresis or heteroduplex analysis; nevertheless, complete sequencing of the globin gene represents the best option to identify rare or unknown mutations.
![]()
CYTOGENETIC DIAGNOSTICS
Metaphase Karyotyping
Traditional cytogenetics, utilizing banding techniques, is performed on chromosomal metaphase spreads. Because mitotic activity is required, metaphase karyotyping is performed after cell culture in the presence of mitogens. For myeloid disorders, either lymphocyte-conditioned media or hematopoietic growth factors are most commonly used, while, for lymphoid malignancies, lectins are added. Various banding methods have been utilized for chromosome identification and resolution of individual chromosomal fragments, but G-banding is usual in clinical diagnostics. Characteristic bands result from the biochemical properties of chromatin such as AT and GC content.4–8
Cellularity and mitotic activity affect the diagnostic yield of the procedure, and the proportion of noninformative spreads varies from disease to disease. In myelofibrosis, marrow is often not aspirable. In aplastic anemia and myelodysplasia, noninformative results are frequent due to the lack of progenitor cells. In such cases, cytogenetic analysis may be also performed on blood specimens.
Approximately 330 chromosomal bands can be distinguished by routine karyotyping, and each band may contain as much as 107 base pairs (bp) and a multitude of genes. Classic karyotyping can identify defects of approximately 5 Mb; thus, smaller defects and their locations may remain undetected (resolution). The sensitivity level depends on the number of analyzed cells; routinely 20 cells are counted with a detailed analysis of at least 2 cells. Analysis may be more complicated if several clones, each harboring a distinct defect, are present. Depending on the nature of the identified defect, the sensitivity limit is approximately 10% (i.e., identification of 2 abnormal cells) in 20 cells tested.
Both balanced and unbalanced translocations can be identified, but some defects may require a more intricate analysis. Some of the balanced translocations are highly diagnostic; examples are t(9;22) in chronic myelogenous leukemia (CML), t(15;17), inv 16 and t(8:21) in acute myeloid lymphoma (AML), t(15:17) in acute promyelocytic leukemia (APL), t(9:22) and t(12:21) in acute lymphoblastic leukemia (ALL), as well as t(14:18), t(11:14), t(11:18) in lymphomas. Once a specific defect is identified, metaphase karyotyping can be used for monitoring of therapy response (cytogenetic remission); however, the sensitivity of this method is limited.5–8
Fluorescence In Situ Hybridization
For the targeted detection of specific abnormalities, fluorescence in situ hybridization (FISH) is the most commonly applied method particularly helpful in the characterization of structural chromosomal abnormalities and identification of chromosomes of uncertain origin. However, FISH is not suitable for screening for unknown defects unless a high clinical suspicion exists. FISH does not require cell division and consequently cell culture, and is more sensitive than traditional cytogenetics. FISH provides a more accurate measure for the true frequency of abnormal cells and can be used for the monitoring of minimal residual disease (MRD). Identification of the donor versus recipient origin of the blood cell production following hematopoietic stem cell transplantation is another application of this technology (see below). The technique can be applied to blood, marrow, body fluids, tissue touch preparations as well as to paraffin-embedded tissues.5,9
In FISH, specific fluorescently labeled single-stranded DNA probes are hybridized to the nuclei of metaphase or interphase cells attached to glass slides. The use of probes labeled with different dyes allows for multicolor FISH on a single slide. Probes can also be designed to identify a specific chromosomal structure, hybridize to multiple chromosomal sequences, and to identify unique DNA sequences. Probes recognizing α-satellite sequences are chromosome specific; in diploid cells both chromosomes are labeled. Chromosome painting probes are derived from whole chromosomes (see also spectral karyotyping [SKY], discussed next). Probes can be derived from unique sequences cloned from specific regions of the genome. Finally, telomeric probes can be used to determine the telomere length based on the intensity of the hybridization.
For balanced translocations, probes spanning individual breakpoints are used. Dual-color/dual-fusion probes or single-fusion/dual-color FISH probes target sequences located at opposite ends of two breakpoints. In addition, two-color break-apart probes, recognizing DNA sequences from the 3´ and 5´ends of a single gene, can be applied. These probes yield combined yellow signal in the normal germline configuration while two colors are seen when target sequences are separated because of translocation. FISH is more reliable for the detection of duplication of chromosome fragments than deletions. In general, FISH is less sensitive than PCR, with detection limits of 1 of 100 cells. As a result of the false-positive rate, it is not clear whether sensitivity can be increased through routine counting of a higher number of cells.
FISH techniques have been widely applied for the detection of lymphoma-specific translocations, in the diagnosis of CML, myelodysplastic syndrome (MDS), and T-cell acute lymphoblastic leukemia (T-ALL) and B-cell acute lymphoblastic leukemia (B-ALL) (Table 29.1).6,8–10 In addition, FISH is frequently used for intracellular detection of Epstein-Barr virus (EBV) in certain non-Hodgkin’s lymphomas, Hodgkin’s disease, and aggressive natural killer (NK) cell lymphomas (see later).
Spectral Karyotyping
SKY allows for the visualization of all 24 chromosomes and analysis of their structure based on hybridization with multicolor painting probes.11
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


