Figure 20.2 shows the histologic appearance of the major soft tissue sarcoma subtypes, and Table 20.1 outlines diagnostic histologic characteristics and molecular and cytogenetic abnormalities.
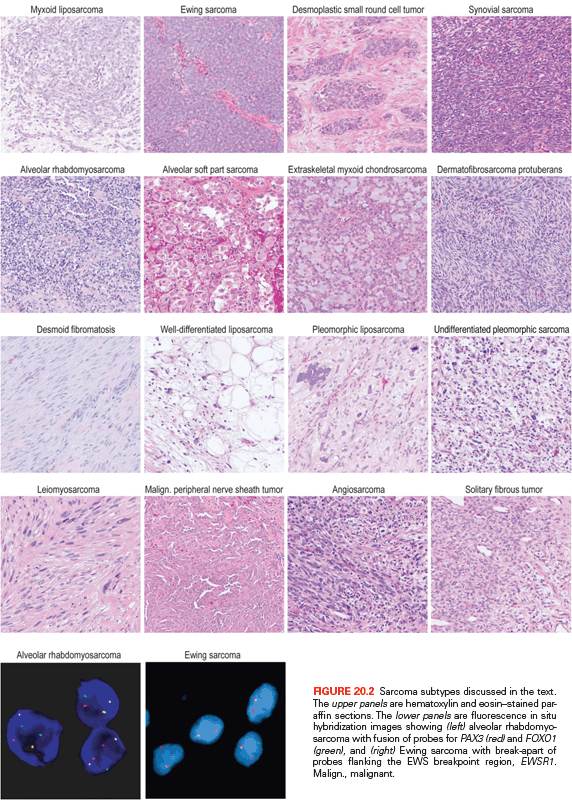
TRANSLOCATION-ASSOCIATED SOFT TISSUE SARCOMAS
Myxoid/Round Cell Liposarcoma
Myxoid liposarcomas typically arise in the thigh or other deep soft tissues in adults (peak age, 30 to 50 years). Myxoid liposarcomas can usually be diagnosed with confidence by its characteristic morphology: a myxoid matrix, a plexiform vasculature, and lipoblasts. These features may, however, be partially lost in its high-grade form, round cell liposarcoma. Nearly all myxoid/round cell liposarcomas carry a balanced translocation, t(12;16)(q13;p11),1 fusing FUS (also known as TLS) with DDIT3 (aka CHOP, GADD153).2 In rare cases, EWSR1 substitutes for its homolog FUS. At least 12 FUS-DDIT3 transcript variants have been reported,3,4 and several are known to induce a sarcoma phenotype in model systems.5,6 The translocations fuse 5′ exons of FUS (encoding transcriptional regulatory domains that interact with the RNA polymerase II complex7) to the full coding sequence of DDIT3, a leucine-zipper transcription factor with roles in cell cycle control,8 adipocytic differentiation,9 and stress response.10 The fusion oncoprotein binds cofactors including C/EBPβ to deregulate gene expression, although few direct targets have been validated to date.11,12 One result is activation of critical pathways, including those related to angiogenesis (interleukin 8 [IL-8]), early adipose differentiation (PPARγ), growth factor signaling (insulinlike growth factor [IGF], RET), and cell-cycle control (cyclin D, CDK4).12–15 Another result is repression of miR-48616 and IL-24,17 which might otherwise act as tumor suppressors.
Clinically, evidence for FUS-DDIT3 translocations from reverse transcription polymerase chain reaction (RT-PCR)3 or fluorescence in situ hybridization (FISH)18 can help confirm the diagnosis and may be useful for small biopsies dominated by a round cell component. Fusion subtype, however, appears to have little prognostic value independent of stage and grade. In general, molecular markers in myxoid liposarcoma have been difficult to test for independent prognostic value, given the difficulty of assembling large series.19 Nevertheless, high levels of p53, IGF1R/IGF2, AXL, and RET may be adverse factors.13,20,21 In addition, mutations in PIK3CA, found in 18% of myxoid/round cell liposarcomas, were associated with a worse outcome.22
Myxoid liposarcomas have dense microvasculature and high expression of IL-812 and vascular endothelial growth factor (VEGF).23 These characteristics suggest a value for antiangiogenic therapies and may underlie the observed sensitivity to radiotherapy24 and trabectedin.25 Trabectedin may also function by disrupting the binding of FUS-DDIT3 to target promoters.11 Agents designed to target FUS-DDIT3 are not yet available.
Ewing Sarcoma
Ewing family tumors appear most commonly in adolescents and young adults; primary sites are most often in bone but can also be in soft tissues. A range of aggressive small blue round cell tumors have been subsumed under the general term Ewing sarcoma family tumor because of common pathognomonic chromosomal translocations.26,27 EWSR1, the common 5′ translocation partner, is fused to one of several ETS family transcription factor genes (usually FLI1).28 In the chimeric protein, EWSR1 provides, at minimum, its N-terminal transcriptional regulatory domain29 and loses its RNA recognition domain. The ETS factor provides its C-terminal DNA-binding domain and loses its native transactivation domain. Several direct transcriptional targets for the fusion oncoprotein are supported by strong evidence. In Ewing sarcoma tissue, some of these targets are upregulated (PTPL1,30 PRKCB,31 DAX1/NR0B132) and some repressed (FOXO1,33 TGFBR2, LOX,34 IGFBP3,35 and the Let-7 microRNA precursor36), but gene repression, mediated by cofactors, appears to predominate.37 The net result is the activation of pathways driving proliferation and cell survival38 and the repression of pathways promoting mesenchymal differentiation.39,40
Molecular confirmation of an EWSR1 translocation can be critical for patient management because many of the clinical, morphologic, and immunophenotypic features of Ewing sarcoma are shared with entities such as mesenchymal chondrosarcoma and small cell osteosarcoma. Commercially available EWSR1 split-apart FISH probes are valuable ancillary diagnostic tools (see Fig. 20.2)41; RT-PCR alternatives are complicated by the need to cover the many alternative fusion sites, but offer the advantage of identifying the specific fusion.42
Several agents (including inhibitors of IGF/mammalian target of rapamycin [mTOR], histone deacetylase, and cyclin-dependent kinase43) that target translocation-induced mechanisms and pathways are currently in clinical trials for Ewing sarcoma, and strategies to inhibit the oncoprotein itself are in active development.43–45
Desmoplastic Small Round Cell Tumor
In desmoplastic small round cell tumors, the same 5′ portions of EWSR1 involved in Ewing sarcomas are fused to WT1,46,47 a tumor suppressor deleted in Wilms tumor.48 The chimeric protein includes the last three WT1 DNA-binding zinc finger domains. Despite some similarities to Ewing sarcoma family tumors, desmoplastic small round cell tumors are rarely cured with aggressive conventional chemotherapy combined with surgical debulking; prognosis is dismal, so new therapies are needed.49,50 Several targets of EWSR1-WT1 have been identified. EWSR1-WT1 directly induces PDGFA expression,51 which explains the desmoplastic background and, along with VEGFA and VEGFR2 overexpression, accounts for the observed partial responses to sunitinib.52 IL2RB is also induced, and its downstream JAK/STAT and AKT/mTOR signaling pathways appear active,46,53,54 representing potential targets for novel treatment approaches.
Synovial Sarcoma
Synovial sarcoma differs from most translocation-associated sarcomas in that the genes involved in its defining translocation, t(X;18)(p11;q11), encode epigenetic regulators, not transcription factors that bind DNA directly.55 The translocation fuses the widely expressed SS18 (aka SYT) gene with an SSX gene normally expressed only in testis.56 In the fusion oncoprotein, SS18, which forms part of the BAF chromatin remodeling complex,57 retains all but the last eight amino acids from its C-terminal transcriptional activation domain. The SSX partner (SSX1, 2, or 4) retains only its C-terminal 78-residue repressor domain, which confers nuclear localization in association with Polycomb proteins.58 The forced expression of SS18-SSX in mesenchymal stem cells59 or conditional expression in mice55,60 recapitulates synovial sarcoma. Neither SS18 nor SSX carries a DNA-binding motif; however, the chimeric oncoprotein gains at least two abnormal functions affecting epigenetic gene regulation. The retained C-terminus of SSX binds TLE1, a highly expressed diagnostic marker of synovial sarcoma61 that recruits Polycomb repressor complex proteins. SS18 binds the transcription factor ATF2, thereby directing the complex to promoters bearing cAMP response elements (CRE). The net result is Polycomb-mediated epigenetic repression of ATF2 target genes, including the tumor suppressors EGR1 and CDKN2A.62 Also, when SS18-SSX replaces native SS18 in BAF complexes, the SMARCB1 (SNF5) component is evicted.63 This latter event increases Polycomb activity and induces hyperactivity of stem cell–associated programs,64 a feature of synovial sarcoma.65 Key genes and oncogenic pathways that become activated, directly or indirectly, in synovial sarcoma include histone deacetylases,62 SOX2,63 Wnt/β-catenin,66 TWIST1,67 FGFR2,68 BCL2,69 and the Akt/mTOR pathway70 via IGF2.59,71,72 Therefore, these represent candidates for targeted therapy approaches in the absence of known drugs that inhibit SS18-SSX directly.
Copy number alterations are more common in adult than in pediatric patients, and both copy number alterations and an expression signature of genes related to mitosis and chromosome function are associated with metastasis.73
Alveolar Rhabdomyosarcoma
In alveolar rhabdomyosarcoma, an aggressive cancer of older children and adolescents, the transcriptional activation domain of FOXO1 (i.e., FKHR) from 13q14 is fused to the DNA-binding domain of paired box transcription factor PAX3 (2q35) or PAX7 (1p36).74,75 Until recently, about 20% of cases were thought to be translocation negative, but in fact, such tumors represent histologic variants of embryonal rhabdomyosarcoma.76 Translocations involving PAX3 may be associated with a worse prognosis than those involving PAX7.77 Thus, to optimize patient care, the diagnosis should be confirmed by FISH (see Fig. 20.2) and/or RT-PCR.78 Either translocation results in a high-level nuclear expression of a chimeric transcription factor that abnormally activates PAX targets, many of which are involved in neurogenesis and are not expressed in normal skeletal muscle.79,80 Recently, PAX3-FOXO1 fusion mRNA and chimeric protein were found to be expressed transiently during normal fetal muscle development (i.e., in cells without the DNA translocations).81 Direct targets of PAX3-FOXO1 include P-cadherin (CDH3),82 GREM1, DAPK1, and MYOD1,83 as well as PDGFRA. Inhibitors of PDGFRA are effective in mouse models.84 The list of targetable kinases expressed in alveolar rhabdomyosarcoma also includes IGF1R and ALK.85–87 Another probable direct target of PAX3-FOXO1 is the cell-cycle regulator SKP2,88 perhaps helping explain why alveolar rhabdomyosarcoma is responsive to conventional cytotoxic chemotherapy.
Alveolar Soft-Part Sarcoma
Alveolar soft-part sarcoma has a clinical presentation and pathognomonic molecular event with many similarities to other translocation-associated sarcomas.89 In this disease, the 5′ half of the widely expressed ASPSCR1 (i.e., ASPL) gene on 17q25 is fused to exon 3 or 4 of TFE3 on Xp11, the latter retaining its transcriptional activation, basic helix-loop-helix, and leucine zipper domains.90 Interestingly, similar fusions are present in a subset of renal cell carcinomas, particularly those arising in younger patients,91 and in a subset of perivascular epithelioid cell neoplasms.92 Although alveolar soft-part sarcoma has distinctive histology, two useful diagnostic adjuncts are the detection of TFE3 rearrangements by FISH93 and the detection of translocations by RT-PCR or by immunohistochemistry for TFE3.94,95 Kobos et al.96 combined expression profiling, chromatin immunoprecipitation, and functional validation to identify a CACGTG consensus motif in genes transcriptionally activated by the ASPSCR1-TFE3 oncoprotein. Their list of validated direct targets97 includes MET and angiogenic mediators, which is consistent with previous studies.97–99 Antiangiogenic therapy is effective in xenograft models.100 A single-arm phase II study of the VEGFR inhibitor cediranib in metastatic alveolar soft-part sarcoma showed a high rate of disease control in association with downregulation of angiogenic genes (including ANGPT2), supporting advanced trials of antiangiogenic agents.101
Dermatofibrosarcoma Protuberans
A hallmark of dermatofibrosarcoma protuberans (DFSP) is supernumerary ring chromosomes that contain material from chromosomes 17 and 22,102–104 or less commonly, an unbalanced der(22)t(17;22)(q21-23;q13). The molecular consequence of both types of aberration is the overexpression of the PDGF beta (PDGFB) gene on chromosome 22, through fusion with the collagen gene COL1A1 on chromosome 17.105,106 The same fusion gene is also seen in two histologic variants: giant cell fibroblastoma and Bednar tumor (pigmented DFSP). FISH and comparative genomic hybridization (CGH) studies have indicated that increased COL1A1–PDGFB copy number is associated with fibrosarcomatous transformation of DFSP, although the copy number increase is not an invariable feature of these cases.107,108
The COL1A1-PDGFB fusion product signals through the PDGF receptor in an autocrine loop.109 This signaling can be blocked using tyrosine-kinase inhibitors acting at PDGFR, such as imatinib. A number of clinical studies have shown a high response rate to imatinib therapy in both locally advanced and metastatic DFSP.102,110,111 These results support the concept that DFSP cells depend on aberrant activation of PDGF signaling for proliferation and survival.
Extraskeletal Myxoid Chondrosarcoma
Most extraskeletal myxoid chondrosarcomas show reciprocal translocations that fuse NR4A3 in 9q22-q31.1 with one of four partners: EWSR1 in 22q12 (the most common), TAF15 in 17q11, TCF12 in 15q21, or TFG in 3q12.112–114 Because these fusion genes have not been described in any other tumor type, they represent useful diagnostic markers. The four different fusion partners have unknown prognostic significance.
NR4A3 encodes a ubiquitously expressed orphan nuclear receptor also known as NOR-1, TEC, MINOR, or CHN.115 The t(9;22) fuses the transactivation domain of EWSR1 to the full length of NR4A3. Analogous to EWSR1-ETS fusions, the EWSR1-NR4A3 fusion protein not only displays strong transcriptional activity, but also regulates RNA splicing.116
TAF15, like EWSR1 and FUS, belongs to the FET family, and contains a characteristic 87–amino acid RNA recognition motif implicated in protein-RNA binding.117 The N-terminal regions of EWSR1, FUS, and TAF15 contain degenerate repeats of the SYGQ motif and mediate powerful transcriptional activation when fused to the heterologous DNA-binding domains of a variety of transcription factors.118
By gene profiling, extraskeletal myxoid chondrosarcomas constitute a distinct genomic entity, showing upregulation of several genes, including NMB, DKK1, DNER, and CLCN3.119 In situ hybridization confirmed that NMB is highly expressed in extraskeletal myxoid chondrosarcoma but not in other sarcoma types, suggesting its potential value as a diagnostic marker.
Solitary Fibrous Tumor and Hemangiopericytoma
Solitary fibrous tumor (SFT) and hemangiopericytoma share similar histopathologic features and are now classified as a single biologic entity.120 In a recent study using paired-end transcriptome sequencing, a recurrent NAB2-STAT6 fusion was identified in virtually all SFTs, regardless of anatomic location (pleura, meninges, or soft tissue).121 In the normal genome, NAB2 and STAT6 are adjacent genes on chromosome 12q13 that are transcribed in opposite directions, but in SFT, the fusion is the result of a chromosomal inversion, fusing NAB2 and STAT6 in a common direction of transcription.
In contrast to the known activity of NAB2 as a repressor of early growth response protein 1 (EGR1) activity, the NAB2-STAT6 fusion induces expression of EGR1 target genes. In an RNA-sequencing analysis of seven SFTs and 282 other tumor samples, the SFTs showed a high-level expression of both EGR1 target genes, including NAB2, NAB1, IGF2, FGF2, and PDGFD, and receptor tyrosine kinases, such as FGFR1 and NTRK1.121 Overexpression of other tyrosine-kinase receptor genes, such as DDR1 and ERBB2, has been seen in array-based profiling.122
IGF2 is uniformly overexpressed in SFT, regardless of anatomic location.122 IGF2 is imprinted on the paternal allele in most adult tissues, and IGF2 overexpression in SFTs was related to a loss of imprinting. Although IGF2 acts by binding to IGF1R, SFTs do not show up-regulation of IGF1R, and it was suggested that IGF2 signaling occurs through the insulin receptor A pathway.123 Overexpression of IGF2 and consequent activation of the insulin receptor may also explain why a subset of SFT patients presents with hypoglycemia. This syndrome, known as Doege-Potter syndrome, has been associated with large tumor size and aggressive clinical behavior and is resolved by surgical resection of the lesion.124
Although a PDGFRB mutation (D850V) was reported in a malignant pleural SFT,125 none of the 39 SFTs tested subsequently showed mutation in this hot spot, nor did they show upregulation of PDGFRB mRNA.122
SOFT TISSUE SARCOMAS OF SIMPLE KARYOTYPE ASSOCIATED WITH MUTATIONS
Desmoid Fibromatosis
Desmoid-type fibromatoses are locally infiltrative, clonal fibroblastic proliferations that arise in the deep soft tissues and never metastasize. Although about 70% result from mutations in adenomatous polyposis coli (APC) or CTNNB1, tumorigenesis may be also be influenced by endocrine and physiologic factors such as pregnancy, trauma, and prior surgery. Desmoids are usually divided into two groups: sporadic desmoids and those in individuals with a heterozygous germ-line mutation in the APC gene (chromosome 5q). Although germ-line APC mutations also often result in familial adenomatous polyposis,126 some desmoid patients harboring such a mutation have no polyposis. The desmoids in individuals with a germ-line APC mutation display inactivation of the second copy of APC, which usually occurs by point mutation or deletion.126,127
Among sporadic desmoids, only a minority display APC inactivation. A majority (52% to 85%) have an activating point mutation in the β-catenin gene, CTNNB1.128,129 These CTNNB1 mutations stabilize β-catenin, resulting in its overabundance. β-Catenin, a mediator of Wnt signaling, is negatively regulated by APC, so both APC inactivation and CTNNB1 activating mutations result in the upregulation of the Wnt pathway. The specific CTNNB1 mutation may have prognostic significance; patients with S45F-mutant desmoids were reported to have a 5-year recurrence-free survival of only 23%, compared with 57% for those with T41A-mutant tumors and 65% for those with wild-type CTNNB1.128 These results raise the possibility that mutation status might aid in selecting patients for more aggressive therapy.
Based on the findings of APC inactivation or activating CTNNB1 mutations in the majority of patients, the development of small-molecule β-catenin antagonists would be likely to provide significant benefit, particularly for patients with advanced disease in whom surgical resection is not feasible. Although such β-catenin–targeted agents are still in preclinical development, an inhibitor of matrix metalloproteinase, a downstream target of β-catenin, substantially reduced tumor volume and tumor invasion in a transgenic Apc+/Apc1638N mouse model of aggressive fibromatosis.130 Hedgehog signaling is activated in human and murine desmoid tumors. Inhibiting Hedgehog signaling reduced cell proliferation and β-catenin protein levels in human desmoid cell cultures and reduced the tumor size and number in a murine model.131 These results suggest Hedgehog antagonists as a promising therapy for desmoid patients.
Patients with desmoids were found to have elevated levels of PDGF-AA and PDGF-BB, leading to a trial of the tyrosine-kinase inhibitor imatinib in patients with advanced disease. Of 19 patients, 3 (16%) had a partial response to treatment and 4 additional patients had stable disease for more than 1 year; overall, the 1-year tumor control rate was 37%.132 The response in these tumors was thought to be mediated by inhibition of PDGFRB kinase activity. Sorafenib, a multitargeted tyrosine-kinase inhibitor, results in tumor shrinkage in 25% of desmoid patients and stable disease in 70%, along with symptom relief in 70% of patients.133
COMPLEX SOFT TISSUE SARCOMA TYPES
Well-Differentiated and Dedifferentiated Liposarcoma
Well-differentiated and dedifferentiated liposarcomas represent the most common biologic group of liposarcoma. This group is characterized by amplification of 12q, which usually occurs in double minutes, ring chromosomes, and large marker chromosomes. In addition, the 12q13.2-q23.1 locus often harbors complex rearrangements (see Fig. 20.1).22,134,135 The amplified region generally includes the oncogenes MDM2, HMGA2, and CDK4, but 12q may contain additional driver genes. On the basis of the rearrangements and correlated overexpression results, possibilities include NAV3, WIF1, MDM1, DYRK2, ELK3, DUSP6, YEATS4, TBK1, and FRS2, which are amplified in ~14% to 80% of tumors. Aside from 12q aberrations, dedifferentiated liposarcomas contain significant amplifications of 1p, 1q, 5p, 6q, and 20q.22,134,135
Amplification of JUN (on 1p32) has been suggested as the explanation for the block in adipocyte differentiation in undifferentiated sarcomas.136 However, JUN is amplified or overexpressed in only a subset of dedifferentiated liposarcomas (DDLS).22,137 Another gene that may be involved in the differentiation phenotype is C/EBPα, which is underexpressed in many DDLS tissues and cell lines. Exogenously expressing C/EBPα resulted in a 50% decrease in proliferation, a G2/M arrest, apoptosis, and restoration of the ability to induce early adipogenesis markers.138 CCAAT-enhancer-binding protein alpha (C/EBPα) expression independently predicts distant recurrence–free survival for patients with primary liposarcoma,139 and loss of the 19q region encompassing C/EBPα is associated with poor outcomes in primary retroperitoneal DDLS.140 In addition, C/EBPα downregulation may result from epigenetic defects; 24% of DDLSs had C/EBPα promoter methylation and 8.3% had somatic mutations in the histone deacetylase HDAC1. Treating DDLS cells with a demethylating agent and the histone deacetylase inhibitor vorinostat increased C/EBPα expression 19-fold, decreased proliferation, induced apoptosis, and reduced xenograft tumor growth by 50% to 70%.141 Taken together, these results suggest that C/EBPα acts as a tumor suppressor in DDLS and that its loss may explain the undifferentiated state of DDLS.
A microarray analysis of gene expression has demonstrated that well-differentiated liposarcomas and DDLS have activation of cell-cycle and checkpoint pathways, including the upregulation of CDK4, MDM2, CDK1, CDC7, TOP2A, PRC1, PLK1, and cyclins B1, B2, and E2.142,143 Therefore, these pathways may be useful as therapeutic targets. In fact, nutlin-3a, a selective MDM2 antagonist, induces apoptosis and inhibits the proliferation of DDLS cell lines at concentrations that do not affect normal adipose-derived stem cells.142 Furthermore, PD0332991, a selective CDK4/CDK6 inhibitor, inhibits proliferation by inducing G1 cell-cycle arrest and senescence in DDLS cell lines and xenografts.22 In a recent phase II trial of PD0332991 in patients with advanced CDK4-amplified liposarcoma, 66% of patients were progression free at 12 weeks, and a subset had a radiographic response and prolonged stable disease.144 These results provide a rationale for use of MDM2 antagonists and CDK4 inhibitors in patients with well-differentiated liposarcomas and DDLSs.
Pleomorphic Liposarcoma
Pleomorphic liposarcoma, accounting for 5% of all liposarcomas, is the least common subtype. It is characterized by high chromosome counts and complex rearrangements, with many unidentifiable marker chromosomes and nonclonal alterations. A high-resolution single nucleotide polymorphism (SNP) array analysis has revealed multiple regions of significant copy number amplification and deletion.145 The most common alteration, found in approximately 60% of tumors, was a deletion of 13q14.2-q14.3, including the RB1 tumor suppressor. The next most common alteration was a loss of 17p13.1, including TP53. Both RB1 and TP53 deletions were a mixture of hemizygous loss and, less frequently, homozygous deletion. In addition, TP53 point mutations were found in 17% of tumors.22 In TP53-mutant cells, antagonism of MDM2 by nutlin-3a enhances chemosensitivity,146 suggesting the potential therapeutic utility of combining nutlin-3a with chemotherapy in TP53-mutant pleomorphic liposarcoma. Small molecules that reactivate mutant TP53, such as PRIMA-1 (APR-246),147 are presently in phase I trials.
A third genetic alteration identified in an SNP analysis was the deletion of 17q11.2, including the tumor suppressor NF1. Among 24 pleomorphic liposarcomas, 9 (38%) had NF1 loss, including 1 case of a homozygous deletion and 2 cases of a mutation of the nondeleted allele.22 Because a loss of NF1 function appears to activate the RAS and mTOR pathways, the frequent NF1 aberrations suggest that MEK or mTOR inhibitors may have clinical utility.
Myxofibrosarcoma and Undifferentiated Pleomorphic Sarcoma (Malignant Fibrous Histiocytoma)
Pathologists now regard myxofibrosarcoma as a distinct tumor type with clearly defined criteria for diagnosis.120,148,149 Undifferentiated pleomorphic sarcoma, however, is less well defined, and it remains controversial whether it represents either (1) a pleomorphic sarcoma showing fibroblastic/myofibroblastic differentiation and thus sharing a common set of genomic alterations with myxofibrosarcoma, or (2) an end-stage undifferentiated morphologic pattern with genomic alterations distinct from those of myxofibrosarcoma.
Myxofibrosarcoma
Myxofibrosarcoma, also known as myxoid variant of malignant fibrous histiocytoma, is a malignant fibroblastic lesion with variably myxoid stroma (at least 10%) composed of hyaluronic acid and solid sheets of spindled and pleomorphic tumor cells. Karyotypes tend to be highly complex, often with multiple numerical and structural rearrangements and with chromosome numbers in the triploid or tetraploid range.150–152 No consistent chromosomal aberration has emerged. In general, karyotype complexity is greater in high-grade lesions and in recurrences.152
In an SNP array analysis of 38 myxofibrosarcomas, approximately 55% harbored chromosome 5p amplification.22 This region contains RICTOR (a binding partner of mTOR), CDH9, and LIFR. Other amplified regions included several discontinuous loci on 1p and 1q spanning PI4KB, ETV3, and MCL1, among others. MCL1, an antiapoptotic gene, was concomitantly overexpressed in these tumors. Myxofibrosarcomas also harbored deletions of tumor suppressors, including CDKN2A/CDKN2B, RB1, TP53, NF1, and PTEN. These events demonstrate an extensive loss of function in several known tumor suppressors.22
Undifferentiated Pleomorphic Sarcoma (Malignant Fibrous Histiocytoma)
Over 50% of soft tissue sarcomas occurring in older adults are histologically pleomorphic and high grade. Most have traditionally been classified as malignant fibrous histiocytoma (MFH).153,154 MFH was originally defined as a malignant pleomorphic spindle cell neoplasm showing fibroblastic and histiocytic differentiation. More recently, pathologists have accepted that this morphology may be shared by a wide range of malignant neoplasms.155 Many sarcomas that were previously classified as pleomorphic MFH, on careful immunohistochemical and histopathologic analyses, revealed a specific line of differentiation and could be reclassified as myxofibrosarcoma (30%), myogenic sarcoma (30%), liposarcoma (4%), malignant peripheral nerve sheath tumor (2%), or soft tissue osteosarcoma (3%), whereas about 30% had no specific line of differentiation or were myofibroblastic.148 The term undifferentiated pleomorphic sarcoma (UPS) is now reserved for pleomorphic sarcomas that show no definable line of differentiation by current technology.
Because of this change in diagnostic criteria, the genetic basis of UPS is difficult to evaluate. Among the more than 60 cases in the Mitelman Database of Chromosome Alterations in Cancer described as storiform or pleomorphic MFH or MFH not otherwise specified, the karyotypes are highly complex. Most have chromosome numbers in the triploid or tetraploid range, but a few are near haploid.156–160 Telomeric associations, ring chromosomes, and dicentric chromosomes are common. In a CGH study of 33 tumors, 25 of which were UPSs as currently defined, numerous copy number changes were found. The most frequent (found in 50% to 65% of tumors) were gains in chromosome 1p (1p33-p32, 1p31, and 1p21), 1q21, and 20q13 and losses in 1q41, 2q36-q37, 10q25-q26, 13q13-q14, 13q14-q21, and 16q12.161 The copy number profiles of the majority of UPSs closely resembled those of leiomyosarcomas161–164 or pleomorphic or dedifferentiated liposarcomas.165–167 Mutations and/or deletions of TP53, RB1, and INK4a have been suggested to be drivers of oncogenesis.162,168–171
Recent work suggests that the stemlike tumor initiating cells (side population cells) isolated from UPS show activation of both the Hedgehog and Notch pathways. Inhibition of these pathways in UPS xenograft models decreased the proportion of side population cells and suppressed tumor self-renewal. This work shows that targeting signaling pathways activated in a small subpopulation of tumor-initiating cells has a dramatic effect on tumor self-renewal and may be a promising approach for these undifferentiated tumors.172
Leiomyosarcoma
Leiomyosarcoma is defined as a malignant tumor with evidence of smooth muscle differentiation. Karyotypes tend to be complex, with amplifications, gains, and losses involving multiple chromosomes.173–175 Frequently observed aberrations include losses of 1p12-pter, 2p, 13q14-q21 (including RB1),176 10q (including PTEN),177 and 16q and gains of 17p, 8q, and 1q21-31; these aberrations have been associated with aggressive clinical behavior. The myocardin (MYOCD) gene on 17p is significantly amplified and overexpressed in retroperitoneal tumors. Knockdown of MYOCD in leiomyosarcoma cell lines harboring this amplification decreases smooth muscle differentiation and inhibits cell migration.178 In an analysis of copy number alterations in 27 leiomyosarcomas,22 deletions, which were more common than amplifications, encompassed known tumor suppressors such as TP53, BRCA2, RB1, and FANCA. The most prominent changes were chromosome 10 deletions (50% to 70% of cases) (see Fig. 20.1). Indeed, genetic inactivation of Pten (human 10q23.21) in smooth muscle in mice recapitulates human leiomyosarcoma,179 suggesting that 10q loss occurs early in leiomyosarcomagenesis. Moreover, partial inactivation of Pten and Tp53 in the smooth muscle lineage in mice results in the development of high-grade pleomorphic sarcomas and leiomyosarcomas with complex karyotypes.180 The sarcomas deficient in both Pten and Tp53 showed upregulated notch signaling and a greater metastatic potential, which could be attenuated by a gamma-secretase inhibitor.180 In addition to PTEN inactivation, we identified homozygous deletions in MTOR. Because PTEN is a repressor of Akt, both these events suggest a critical role for aberrant Akt-mTOR signaling in leiomyosarcoma. mTOR inhibitors such as everolimus (RAD001) and temsirolimus have shown some efficacy in patients with leiomyosarcoma in clinical trials.181,182
RB1 deletion is common in leiomyosarcomas, with 70% harboring heterozygous deletions and 8% harboring homozygous deletions. A role for RB1 deletion in leiomyosarcoma fits with the high incidence of leiomyosarcoma in individuals with hereditary retinoblastoma.183
Malignant Peripheral Nerve Sheath Tumor
Malignant peripheral nerve sheath tumors (MPNST) are highly aggressive soft tissue sarcomas that rarely occur in the general population, but are much more common in patients with neurofibromatosis type 1 (NF1), a hereditary tumor predisposition syndrome caused by heterozygous mutations of the NF1 gene. The MPNSTs in NF1 patients typically arise from neurofibromas. NF1 patients’ lifetime risk of developing MPNST is 8% to 13%, contrasting with the 0.001% risk in the general population.184,185
The NF1
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


