In addition to pathway-based analyses, prostate cancer can be defined by recurrent lesions of single genes. Approximately 50% of prostate-specific antigen (PSA)-screened prostate cancers harbor gene fusions involving E26 transformation specific (ETS) transcription factors, which represent oncogenic drivers.8,9 Strikingly, ETS fusions and certain other lesions, such as mutation of the E3 ubiquitin ligase SPOP and fusions involving RAF, appear to be mutually exclusive, suggesting a framework for molecular subtyping of prostate cancer.3,10,11 The sections that follow further describe the altered pathways and recurrent genetic lesions most validated in prostate cancer.
ANDROGEN RECEPTOR BIOLOGY AND THERAPY
Introduction to the Androgen Receptor
AR has remained one of the most important and most studied proteins in prostate cancer. The dependence of most prostate cancers on AR activation has served as a basis for important therapeutic approaches, such as luteinizing hormone releasing hormone (LHRH) analogs, antiandrogens, and androgen biosynthesis inhibitors.
AR is a 110-kb steroid receptor transcription factor, located on Xq12.12 Upon binding to an androgen, AR mediates the transcription of a number of genes involved in survival and differentiation of prostate epithelial cells, starting at development of the normal gland. Expression of AR is high in the luminal epithelial cells but lower in the basal epithelial cells that define glandular structure.13
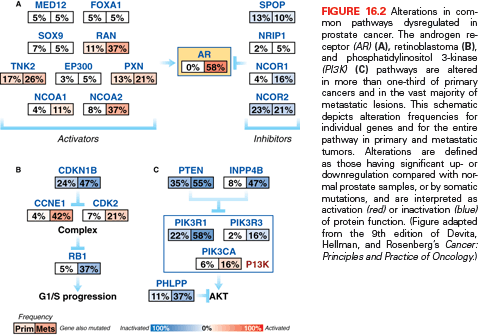
In conjunction with NKX3.1 and FOXA1, AR plays a critical role in normal prostate organogenesis and disease progression.14 NKX3.1 is an AR-regulated prostatic tumor suppressor gene that is located on chromosome 8p and present through development.15 It is the earliest marker of organogenesis and is involved with ductal morphogenesis and secretory function.16 Mutations are infrequent in human cancers, but loss of heterozygosity in 8p becomes more frequent during cancer progression, with 86% loss in prostate cancers (see Fig. 16.1).17 Loss of NKX3.1 has been associated with activation of the TMPRSS2-ERG gene fusion, which promotes tumorigenesis.18 FOXA1 is a member of the forkhead transcription factor family that opens compacted chromatin to facilitate AR recruitement.19 It is known to play a pivotal role in AR and steroid receptor function.20 It is now recognized that mutation and overexpression of FOXA1 can promote progression to CRPC (see Fig. 16.2).3,5
Polymorphisms of AR as well as CYP17 and SRD5A2 are low penetrant risk factors for prostate cancer development in some but not all studies.21 AR-pathway alterations exist in 60% of primary tumors and nearly 100% of metastatic tumors. NCOA2 (also called SRC2) is the most commonly altered member of this pathway (see Fig. 16.2) and is an AR coactivator that potentiates transcriptional output. Several other AR coactivators, including NCOA1, TNK2, and EP300, are upregulated in metastatic disease, whereas AR corepressors, including NRIP1, NCOR1, and NCOR2, are downregulated (see Fig. 16.2).
Targeting Androgen Receptor Activity: GnRH-Targeting Therapies and Androgen Biosynthesis Inhibitors
The inhibition of androgen synthesis has been used a means of suppressing AR activity. Gonadotropin-releasing hormone-luteinizing hormone (GnRH) agonists or antagonists represent the first-line approach for inhibiting androgen synthesis (Fig. 16.3). These therapies work by suppressing the hypothalamic–pituitary–gonadal axis by eventually causing luteinizing hormone (LH) levels to fall, thereby minimizing testicular production of testosterone and its subsequent conversion to dihydrotestosterone (DHT) by 5-alpha reduction. DHT is the most potent activator of AR and binds to the AR, resulting in translocation to the nucleus and activation of transcription (see Fig. 16.3).
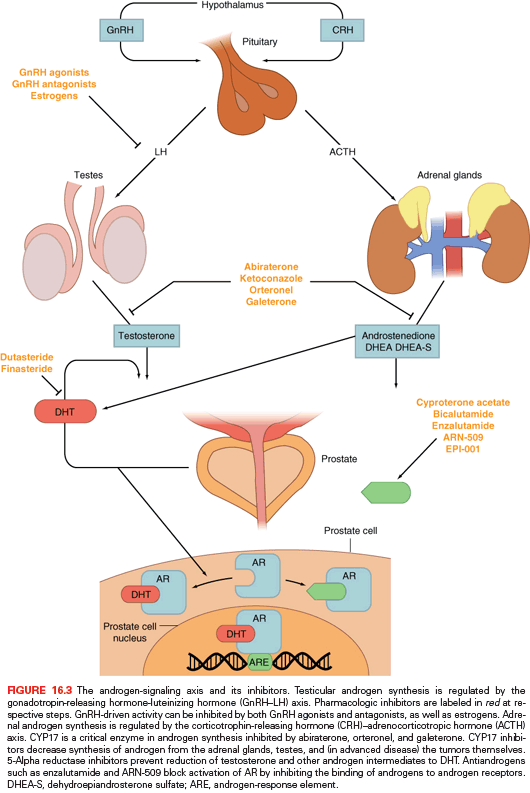
These strategies, although initially highly effective, are often overcome during tumor progression. Alternative sources of DHT and alternative means of AR activity have been identified as mechanisms of resistance. GnRH-targeting agents have no impact on nongonadal androgen synthesis (i.e., suppression of adrenocorticotropic hormone [ACTH] and, hence, adrenal androgen synthesis) (see Fig. 16.3). The adrenal glands can synthesize adequate levels of androgens to promote cancer growth. CYP17 is a key P450 enzyme in the androgen biosynthesis pathway that generates dehydroepiandrosterone (DHEA) and androstenedione in the adrenal glands. These weak androgens can be further converted into testosterone or alternative steroid substrates that are reduced to DHT.22,12 This step is recognized as a particular bottleneck in androgen production. The specific inhibition of CYP17 decreases androgen synthesis, with less effect on the production of other essential steroids. Abiraterone acetate is a pregnenolone derivative that is a high-affinity, irreversible inhibitor of CYP17, and results in decreased adrenal androgen synthesis. At higher doses, this agent inhibits other DHT synthetic pathways such as 3β-hydroxysteroid dehydrogenase.23
Intratumoral androgens may be increased in CRPC.24 Measurements of intratumoral androgens showed that CRPC tumors have more testosterone than primary tumors in untreated men.25 Expression profiling studies comparing CRPC with primary tumors have shown that androgen synthesis enzymes are upregulated in CRPC.26,27 Patient samples at all stages of disease contain an abundance of AKR1C3 and SRD5A1, which are necessary for the conversion of androstenedione to DHT. However, the samples lacked high expression of enzymes necessary for de novo steroidogenesis.28 These data suggest that autocrine androgen synthesis may allow tumors to grow despite low serum androgen levels, but that this process may, in part, be dependent on adrenal precursors.
Targeting Androgen Receptor Activity: Antiandrogens
Antiandrogens compete with endogenous androgens for the ligand binding pocket of AR (see Fig. 16.3). Older agents promote translocation of AR from the cytoplasm into the nucleus and DNA binding, but induce conformational changes that prevent optimal transcriptional activity. In the United States, there have been three historic nonsteroidal antiandrogens (NSAA): flutamide, bicalutamide, and nilutamide. Each has been associated with an antiandrogen withdrawal effect, whereby treated patients experiencing disease progression on treatment derive clinical benefit when the antiandrogen is stopped.29 Although this was initially believed to be related to AR mutation, later studies have shown this is due to AR activation by these NSAAs.30,31
Newer NSAAs have now been developed that lack agonist effects on the AR. Enzalutamide (MDV3100) was approved by the U.S. Food and Drug Administration (FDA) in 2012.32 Other next-generation NSAAs are in development, including ARN-509.
Finally, new approaches are being taken to inhibit AR at the amino terminus.33 As opposed to interrupting ligand binding, agents such as EPI-001 interfere with transcriptional activity of AR. These agents are still in early development, but appear very promising because they display activity even when the ligand-binding domain of AR has been altered.
Alterations in the Androgen Receptor During Prostate Cancer Progression
Prostate cancers eventually progress to a more lethal state: castration resistance. The time to development of resistance and the molecular pathways to CRPC differ from patient to patient. Progression of CRPC is usually accompanied by the restoration of PSA secretion, indicating a reactivation of AR activity in these cases.12
Gene amplification and increased protein expression represent the most common mechanisms of means of reactivating AR signaling in the face of castration therapy. These changes allow cancer cells to respond to subphysiologic concentrations of androgen.34 AR mutations are detected in up to 10% of CRPC cases and allow for activation by alternative ligands.35 Overexpression of AR is seen in approximately 30% of cases of CRPC.36 Alternative splicing of AR mRNA can lead to a truncated protein missing the ligand-binding domain. These increase in number in the face of many second-line NSAAs.37,38 Some of these variants have constitutive transcriptional activity in the absence of androgens. Overexpression of these variants can confer castration resistance in preclinical models.39 More recent studies show that these variants impact sensitivity to taxanes40 and even next-generation antiandrogens such as enzalutamide.41
ADDITIONAL PATHWAYS FREQUENTLY ALTERED DURING DISEASE PROGRESSION
The Phosphatidylinositol 3-Phosphate Kinase (PI3K) Pathway
Progression to CRPC is characterized by gain of function in AR and activation of the phosphatidylinositol 3-phosphate kinase (PI3K) pathway. Cross-talk between these pathways has been proposed42,43 because a loss of PTEN and subsequent PI3K activation is associated with repression of androgen responsive genes. Conversely, AR inhibition results in the upregulation of PI3K activity.42,43 As such, combination therapy with PI3K and AR inhibition are currently being evaluated in clinical trials.
The PI3K signaling cascade is one of the most commonly altered pathways in human malignancy. PTEN is a tumor suppressor that deactivates PI3K signaling. PTEN deletions are present in nearly 30% of primary prostate cancers, and inactivating mutations of PTEN occur in another 5% to 10%; both are more common in advanced disease (see Fig 16.2).3,5,7,44,45 Functional studies across multiple preclinical model systems of prostate cancer consistently reinforce the role of PTEN in suppressing tumorigenesis and prostate cancer progression.46,47 Dysregulation of PTEN represents a poor prognostic factor. There is significant evidence that PTEN deletion is associated with higher stage, higher Gleason grade, and higher rates of progression, recurrence after therapy, and disease-specific mortality.48,49
Amplification and point mutations in PIK3CA, encoding a catalytic subunit of PI3K, also result in overactivation of the pathway and are enriched in metastatic versus localized prostate cancer (see Fig 16.2).3,50 Activating PIK3CA lesions and inactivation of PTEN are usually mutually exclusive, supporting a similar endpoint in driving downstream signaling.
Recent studies have also identified the presence of rarer events that affect PI3K signaling. These lesions include the rearrangement of MAGI2, the deletion of PHLPP1, mutations in GSK3B, and point mutations and genomic deletions of CDKN1B.3,4,44,47,51 Recurrent alterations in multiple nodes along the PI3K pathway emphasize its critical importance in the pathogenesis of prostate cancer and support the rationale for therapies targeting this pathway.
The Retinoblastoma Pathway
Another tumor suppressor pathway that is frequently inactivated in epithelial malignancies is the RB pathway (see Fig. 16.2). The p130 RB protein regulates cell cycle progression by binding to E2F family members and repressing E2F-mediated gene transcription. RB is inactivated via phosphorylation by cyclin-dependent kinases (CDK), resulting in E2F-mediated cell cycle progression.52 The absence of RB signaling results in aberrant cell cycle progression and tumor proliferation.
Similar to PTEN deletion, the loss of RB is enriched in prostate cancer metastases compared to primary disease (see Fig. 16.2).53 Within preclinical prostate cancer models, the inactivation of RB confers castration-resistant tumor growth via E2F1-mediated activation of AR gene transcription.53 RB status is currently being explored as a stratification variable for treatment strategies on ongoing clinical trials.
E26 TRANSFORMATION SPECIFIC (ETS) GENE FUSIONS AND THE MOLECULAR SUBTYPES OF PROSTATE CANCER
E26 Transformation Specific (ETS) Gene Fusions
In addition to pathway-based analyses, prostate cancer can be defined by recurrent lesions of single genes. The most common molecular abnormality in prostate cancer is a gene fusion involving an ETS transcription factor.8,9 These genetic rearrangements, found in approximately 50% of prostate cancers, occur when genes encoding an ETS transcription factor are translocated downstream of the regulatory, usually androgen-responsive, element of a second gene.8,9,54 These fusions result in AR-driven overexpression of an ETS transcription factor, causing transcriptional dysregulation of downstream oncogenic pathways, increased invasion, and carcinogenesis in preclinical models of prostate cancer.8,9,54,55 The most prevalent ETS gene rearrangement involves either chromosomal deletion or insertion of chromosome 21, resulting in fusion of the 5′ untranslated region of the androgen-regulated gene TMPRSS2 with the ETS family member ERG. Over 90% of ETS rearrangements involve ERG, whereas the remaining ETS fusions include ETV1 (chromosome 7), ETV5 (chromosome 3), or ETV4 (chromosome 17) as common 3′ partners (Fig. 16.4). Although TMPRSS2 is the most common 5′ partner for ERG, over 10 androgen-regulated genes, including SLC45A3 and NDRG1, have been identified as 5′ fusion partners in ETS rearrangements (see Fig. 16.4).
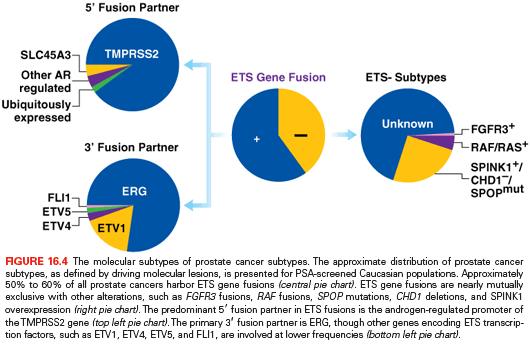
Several studies have investigated the prognostic value of ETS fusion status, with conflicting results likely stemming from the heterogeneity of study cohorts, screening practices, and management strategies, as well as the variability in tumor sampling, disease multifocality, and measured clinical outcomes. In the context of non–PSA-screened populations managed conservatively, population-based studies have demonstrated a significant association between ETS fusion status and adverse clinicopathologic predictors, metastases, or disease-specific mortality.56–58
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


