The most common cancer type of pancreatic cancer is pancreatic ductal adenocarcinoma (PDA). PDA is the primary focus of clinical and molecular research and is thus highlighted in this chapter. Other clinically and molecularly distinct forms of cancer occur in the pancreas and must be distinguished from PDA; they are discussed here in lesser detail.
In recent years, techniques were developed to sequence all of the genes of individual cancers. Whole-exome sequencing of PDAs revealed an average of 63 somatic mutations per tumor.1 Most of these mutations undoubtedly are nonfunctional passenger mutations, each mutated at a low frequency and not contributing to tumorigenesis. Indeed, most passenger mutations might arise as tissues age before tumorigenesis even begins.2 Smoking is associated with a doubling of the risk for pancreatic cancer, and remarkably, it is also associated with a 40% increase of the prevalence of low-frequency mutations in the cancers.3 Only a subset of the mutations in PDA, however, are responsible for driving the neoplastic process in the ducts; only they are discussed further here. Modeling of a comprehensive study mapping gene mutations in multiple regions of primary PDAs and their metastases indicated a general timeline of tumorigenesis, invasion, and metastasis. According to this model, about a decade of time passes between the first driver mutation initiating the precursor neoplasm and the emergence of the first cell having the genotype of the invasive cancer; metastatic ability is acquired after another 5 years, and with patient death following about 2 years later.4
Telomere abnormalities and manifestations of chromosome instability are the most common alterations in pancreatic neoplasia. Four genes are mutated in most PDAs: the KRAS, p16/CDKN2A, TP53, and SMAD4/DPC4 genes. Other recurrent genetic abnormalities are seen at a lower frequency, including internal deletions of exons of FAM190A/CCSER1; mutations in the genes BRCA2, PALB2, FANCC, FANCG, FBXW7, BAX, and RB1, in the transforming growth factor beta (TGF-β) receptors TGFBR1 and TGFBR2, in the activin receptors ACVR1B and ACVR2, in various chromatin-remodeling genes such as ARID1A, in the genes MKK4, STK11, MLL3, ATM, GUCY2F, NTRK3, and EGFR, and in cationic trypsinogen; alterations in the mitochondrial genome; amplifications; various chromosomal deletions; inactivation of DNA mismatch-repair genes; and rarely, the maintenance of the Epstein-Barr virus genome as an episome. Irregular sizes and numbers of centrosomes were observed in 85% of pancreatic cancers and some adenomas, but in no tissues of chronic pancreatitis or normal pancreas.5
Knowing the genes that are mutated in a cancer can have direct clinical impact. For example, some patients develop pancreatic cancer because of an inherited mutation, and these patients and their families could benefit from genetic counseling.6–9 A distinct morphologic subtype of pancreatic cancer, the medullary cancer, can suggest such an inherited mutation.10,11 Another example of clinical impact includes the analysis of the genetic alterations in precursors to invasive pancreatic neoplasia, which indicated that most carcinomas arise by a process of progressive intraductal tumorigenesis, suggesting that these intraductal lesions might be detected and treated before a patient develops an invasive cancer.12 Epigenetic changes in DNA methylation and in gene expression are also highly specific for the cancerous cells and can serve as markers of disease.
COMMON MOLECULAR CHANGES
Telomere shortening is an early and prevalent genetic change identified in the pancreatic precursor lesions.13 Telomere shortening experimentally predisposes cells to chromosome fusion (translocations) and the missegregation of genetic material during mitosis.14 Later in tumorigenesis, the telomerase is often reactivated,15,16 moderating the telomere erosive process while permitting continued chromosomal instability.17
The KRAS gene mediates signals from growth factor receptors and other signaling inputs (Fig. 11.1). The mutations present in most pancreatic cancers convert the normal Kras protein (a protooncogene) to an oncogene, causing the protein to become overactive in transmitting growth factor–initiated signals.18 KRAS is mutated in over 90% of conventional pancreatic ductal carcinomas.19 Among the first genetic changes in the ducts is a KRAS gene mutation (see Table 11.1),20,21 and recent evidence from advanced gene sequencing techniques indicates that their prevalence in early lesions is higher than was previously thought.22
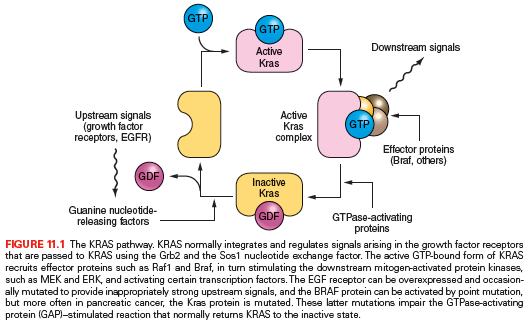
As one of the most commonly mutated genes in pancreatic cancer, the Ras protein is an attractive target for the development of gene-specific therapies, and an understanding of the normal biology of the Ras protein should help in the development of these Ras-targeted therapies. Ras proteins require an attachment to the plasma membrane for activity. For many proteins, including Ras, a hydrophobic prenyl group is essential for the attachment. Either farnesyl (15-carbon) or geranylgeranyl (20-carbon) makes a covalent thioether linkage at a cysteine residue located near the C-terminal end of Ras proteins, termed the CAAX motif. Working mostly in artificial legacy models of the HRAS oncogene (rather than the more widely available but experimentally less tractable natural KRAS-mutant cancer cell lines), the farnesylation reaction was readily inhibited by various means; in these models, the Ras protein was rendered inactive and was often accompanied by cytotoxicity limited to the mutant cells.
Although many types of compounds capable of blocking the farnesyltransferase enzyme were developed as drugs, they have not been successful anticancer agents. Reasons are many. Although the Hras protein is linked predominantly through farnesyl groups, the Kras protein can be alternately prenylated by geranylgeranyl linkages. The latter is thought to be critical for a wider number of cellular proteins, and for fear of excessive toxicity, geranylgeranyl linkages have not usually been considered as an attractive drug target. The Kras protein may bind more tightly than Hras to the farnesyltransferase enzyme, necessitating higher drug concentrations.18 Additionally, the artificial models usually employed the engineered overexpression of the Ras protein, a situation in which the unattached Ras proteins would serve as a dominant-negative inhibitor, binding the essential interacting proteins and sequestering them in the cytoplasm to ensure the inactivation of the many downstream Ras pathways. Such a concentration-driven mechanism would presumably not occur under the normal levels of Ras proteins present in human cancers.23 Indeed, it is proposed that the limited efficacy of farnesyltransferase inhibitors (FTI) observed in some experimental models and in clinical trials may be attributable to a cellular target not yet identified.24 Attention has turned to compounds that target the downstream mediators, such as Raf and Mek protein kinase inhibitors.
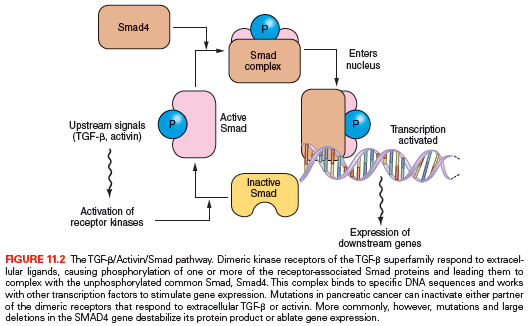
The Smad pathway mediates signals initiated upon the binding of the extracellular proteins TGF-β, activin, and bone morphogenic proteins to their receptors (Fig. 11.2). These signals are transmitted to the nucleus by proteins of the Smad family of related genes, including SMAD4 (DPC4).25 Once in the nucleus, Smad protein complexes bind specific recognition sites on DNA and cause the transcription of certain genes.26 Mutations in the SMAD4 gene are found in nearly half of pancreatic carcinomas, including homozygous deletions or intragenic mutations combined with loss of heterozygosity (LOH).27 Other Smad genes are also occasionally mutated in pancreatic cancer.1 Also, homozygous deletions and mutation/LOH affecting the TGF-β receptor genes are seen in a few pancreatic cancers.28 A more common abnormality, in pancreatic as well as in other tumor types, is the underexpression of TGF-β receptors, which may render cells resistant to the normal suppressive effects of the TGF-β ligand.29
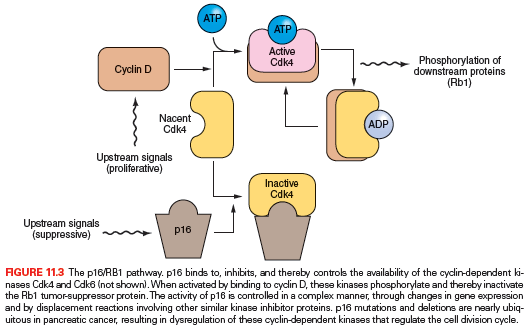
The p16/RB1 pathway is a key control of the cell division cycle (Fig. 11.3). The retinoblastoma protein (Rb1) is a transcriptional regulator and regulates the entry of cells into S phase. A complex of cyclin D and a cyclin-dependent kinase (Cdk4 and Cdk6) phosphorylates and thereby regulates Rb1. The p16 protein is a Cdk-inhibitor that binds Cdk4 and Cdk6.30–32 Virtually all pancreatic carcinomas suffer a loss of p16 gene function through homozygous deletions, a mutation combined with LOH, or promoter methylation of the p16/CDKN2A gene associated with a lack of gene expression.33,34 In addition, inherited mutations of the p16/CDKN2A gene cause a familial melanoma/pancreatic cancer syndrome known as familial atypical multiple mole melanoma (FAMMM).35–39 Only rare pancreatic cancers have an inactivating mutation of the RB1 gene.40
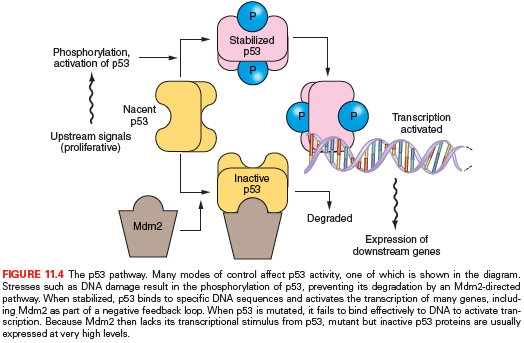
The protein product of the TP53 gene, Tp53, binds to specific sites of DNA and activates the transcription of certain genes that control the cell division cycle and apoptosis.41,42 The Tp53 protein, normally a short-lived protein, becomes phosphorylated and stabilized after DNA damage and other cellular stresses (Fig. 11.4). In about 75% of pancreatic cancers, the TP53 gene has point mutations that inhibit the ability of p53 to bind DNA or, occasionally, other types of inactivating mutation.43–45
Most human carcinomas have chromosomal instability (CIN), which produces changes in chromosomal copy numbers or aneuploidy.46 Most pancreatic cancers have complex karyotypes, including deletions of whole chromosomes and subchromosomal regions.47–49 CIN is the process that causes most of the tumor deletions (i.e., LOH).50 A few percent of pancreatic carcinomas, however, do not have significant gross or numerical chromosomal changes and instead have a different form of genetic instability; they have defects in DNA mismatch repair, producing high mutation rates at sites of simple repetitive sequences (microsatellites), termed microsatellite instability (MSI).10,11,51–54 The pattern of genetic damage in these carcinomas differs considerably from the pattern in carcinomas with CIN. The type II TGF-β and activin receptors (TGFBR2) and (ACVR2), as well as the BAX gene, have a repetitive sequence within their protein-coding regions, and biallelic inactivating mutations of these sequences are seen in many MSI pancreatic cancers.28,54–57
There are also alterations in pancreatic carcinomas, some probably being important to tumorigenesis, that are not attributed to genetic mutations. These include the expression of telomerase,15,16 the underexpression of TGF-β receptors,29 and the overexpression of the growth-stimulating Her-2/neu cell surface receptor58–61 and growth factor-related proteins.62 Some of these activities are proposed to be attractive as therapeutic targets, although supportive clinical evidence is not yet available. Pancreatic carcinomas also have reproducible alterations in gene expression, such as overexpression of the proteins mesothelin and prostate stem cell antigen (PSCA), that currently can serve as diagnostic aids in the histopathologic interpretation of biopsies and surgical resections.57–59 The epigenetic patterns of gene hypermethylation and various patterns of overexpression of RNA transcripts and proteins in pancreatic cancers are considered promising for developing additional diagnostic markers for the analysis of pancreatic secretions and for noninvasive diagnostic screening.63
Genetic patterns are also found among other diagnostic categories of pancreatic neoplasia. The precursor lesions of PDAs, termed pancreatic intraepithelial neoplasia (PanIN), in their most advanced grade closely resemble the genetic patterns of the conventional invasive PDAs. However, the lesions other than the PDAs, including the intraductal papillary mucinous neoplasms (IPMN), the mucinous cyctic neoplasms (MCNs), acinar cell carcinomas, well-differentiated neuroendocrine tumors, pancreatoblastomas, and solid pseudopapillary neoplasms (SPNs), diverge significantly from the patterns of PanINs and typical invasive PDAs. These differences could be used in the future for differential diagnosis of lesions upon biopsy.
Cystic neoplasms such as IPMNs, serous cystadenomas (SCAs), MCNs, and SPNs appear to have relatively few mutant genes in each tumor, yet the genes involved are remarkably distinct.64–66 Mutations in GNAS or KRAS were found in 96% of IPMNs; those in RNF43 were present in many IPMNs and MCNs; those in CTNNB1 were ubiquitous among the SPNs; and those in VHL affected half of the SCAs. GNAS mutations were found in collected pancreatic secretions samples of two thirds of familial and sporadic cases of IPMNs, but not in controls, and their presence predicted subsequent emergence or increasing size of detected cysts.67 PIK3CA mutations are present in some IPMNs and the colloid carcinomas that can derive from them.68 CTNNB1 (beta-catenin) mutations are present in virtually all solid-pseudopapillary neoplasms69 and pancreatoblastomas.70 These genetic data suggested utilities in detection, diagnostic classification, and prognostication when clinically managing pancreatic cysts.
Mutations of MEN1 and either the DAXX or ATRX genes are found in most well-differentiated pancreatic neuroendocrine tumors (PanNET).71,72 Nearly two thirds of these tumors (a subset uniformly harboring DAXX or ATRX mutations) were found to have abnormal telomeres, indicating an active process termed alternative lengthening of telomeres (ALT). ALT is distinct from the typical activation of telomerase implicated in most types of cancer.73 The distinguishing patterns of mutations also firmly establish that high-grade small- and large-cell neuroendocrine carcinomas of the pancreas, despite having histologic similarities with well-differentiated PanNETs, arise through different tumorigenic mechanisms.74
Lower-Frequency Genetic Changes
The causative genes of Fanconi anemia play a role in human tumorigenesis. The BRCA2 gene represents Fanconi complementation group D1 and is thought to aid DNA strand repair.75 Because of this function, it is perhaps best to categorize BRCA2 as a genome-maintenance gene rather than a conventional tumor-suppressor. As many as 7% of apparently sporadic pancreatic cancers (more, in instances of familial aggregation) harbor an inactivating intragenic inherited mutation of one copy of the BRCA2 gene, accompanied by LOH.1,2,66 The PALB2 gene represents Fanconi group N, and its protein product functions by binding the Brca2 protein.76 Three percent of familial pancreatic cancers harbored a germ-line inactivating mutation of PALB2, and in a tumor studied in depth, the other copy was inactivated by a somatic mutation.1,77,78
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


