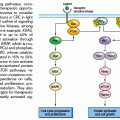
GCs are highly dynamic structures in which B cells transit back and forth between two zones that are conserved across several species: the dark zone (DZ), which consists of rapidly proliferating centroblasts (CB) (doubling time: 6 to 12 hours), and the light zone (LZ), which consists of more quiescent cells termed centrocytes (CC), amidst a network of resident accessory cells (follicular dendritic cells [FDC] and human T follicular helper [Tfh] cells).6–9 According to currently accepted models, the DZ is the site where GC B cells modify the variable region of their IG genes (IgV) by the process of somatic hypermutation (SHM), which introduces mostly single nucleotide substitutions with few deletions and duplications in order to change their affinity for the antigen.3,5,10–12 Conversely, the LZ is the site of selection based on affinity to the antigen. A critical regulator of the GC reaction is BCL6,13,14 a transcriptional repressor15 that negatively modulates the expression of a broad set of genes, including those involved in B-cell receptor (BCR) and CD40 signaling,16,17 T-cell mediated B-cell activation,16 induction of apoptosis,16,18 response to DNA damage (by modulation of genes involved in the sensing and execution of DNA damage responses),19–22 various cytokine and chemokine signaling pathways (e.g., those triggered by interferon and transforming growth factor beta [TGFβ]),16,18 and plasma cell differentiation, via suppression of the PRDM1/BLIMP1 master regulator.23–26 This transcriptional program suggests that BCL6 is critical to establish the proliferative status of CBs and to allow the execution of antigen-specific DNA modification processes (SHM and class-switch recombination) without eliciting responses to DNA damage; furthermore, BCL6 keeps in check a variety of signaling pathways that could lead to premature activation and differentiation prior to the selection for the survival of cells producing high affinity antibodies.
CBs are then believed to cease proliferation and shuttle to the LZ, where they are rechallenged by the antigen through the interaction with CD4+ T cells and FDCs.3,4,7,8 CCs expressing a BCR with reduced affinity for the antigen will be eliminated by apoptosis, whereas a few cells with greater affinity will be selected for survival and differentiation into memory cells and plasma cells,4 or reenter the DZ following stimulation by a variety of different signals.9 Iterative rounds of mutations and selections lead to affinity maturation at the population level. In the GC, CCs also undergo class-switch recombination (CSR), a DNA remodeling event that confers distinct effector functions to antibodies with identical specificities.27 SHM and CSR represent B-cell–specific functions that modify the genome of B cells via mechanisms involving single- or double-strand breaks and which depend on the activity of the activation-induced cytidine deaminase (AID) enzyme,28–30 a notion that will become important in the understanding of the mechanisms generating genetic alterations in B-NHL.
Once these processes are completed, two critical signals for licensing GC exit are represented by engagement of the BCR by the antigen and activation of the CD40 receptor by the CD40 ligand present on CD4+ T cells. These signals induce the downregulation of BCL6 at the translational and transcriptional level, respectively, thus restoring DNA damage responses, as well as activation and differentiation capabilities.
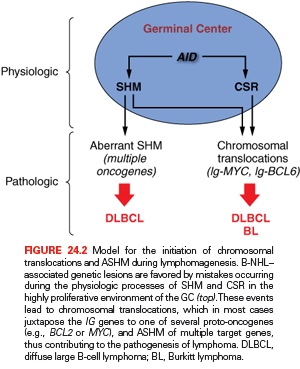
Although oversimplified, this schematic description of the GC reaction is useful to introduce two basic concepts for the understanding of B-NHL pathogenesis. First, the activity of SHM, which introduces irreversible DNA changes in the genome, allowed for the conclusion that most B-NHL types, with the exception of most mantle-cell lymphoma (MCL), derive from GC-experienced B cells that underwent clonal expansion within the GC, because the malignant clones harbor hypermutated IgV sequences containing largely identical mutations, suggesting the derivation from a single founder cell.31 Second, two common mechanisms of oncogenic lesions in B-NHL—namely, chromosomal translocations and aberrant somatic hypermutation (ASHM)—result from mistakes in the machinery that normally diversifies the Ig genes during B lymphocytes differentiation, further supporting the GC origin of most B-NHL (Fig. 24.2).32 Finally, the definition of two distinct phases during GC development reflects different transient states within the same B-cell developmental step, which can be recognized in different B-NHL subtypes to some extent.
T-Cell Development
The process of T-cell development proceeds through sequential stages defined according to the expression of the molecules CD4 and CD8. Committed lymphoid progenitors exit the bone marrow and migrate to the thymus as early T-cell progenitors or double negative 1 (DN1) cells, which lack the expression of both CD4 and CD8 and harbor unrearranged T-cell receptor (TCR) genes.33 In the thymic cortex, T cells advance through the double negative stages DN2, DN3, and DN4, while undergoing specific rearrangements at the TCRβ locus in order to acquire expression of the pre-TCR.33 Those thymocytes that have successfully recombined the pre-TCR will be selected to further differentiate into double positive cells (CD4+CD8+), which express a complete surface TCR and can then enter a process of positive and negative selection in the medulla, before exiting the thymus as single positive T cells.33 The end result of this process is a pool of mature T cells that exhibit coordinated TCR and coreceptor specificities, as required for effective immune responses to foreign antigens. Most mature T-NHLs arise from postthymic T cells in the lymphoid organs.
GENERAL MECHANISMS OF GENETIC LESIONS IN LYMPHOMA
Analogous to other cancers, lymphoma represents a multistep process deriving from the accumulation of multiple genetic lesions affecting oncogenes and tumor suppressor genes, including chromosomal translocations, point mutations, genomic deletions, and copy number (CN) gains/amplifications.
Chromosomal Translocations
Although also found in nonlymphoid tumors, chromosomal translocations represent the genetic hallmark of malignancies derived from the hematopoietic system. These events are generated through the reciprocal and balanced recombination of two specific chromosomes and are often recurrently associated with a given tumor type, where they are clonally represented in each tumor case.
The precise molecular mechanisms underlying the generation of translocations remain partially unclear; however, significant advances have been made in our understanding of the events that are required for their initiation.34 It has been documented that chromosomal translocations occur at least in part as a consequence of mistakes during Ig and TCR gene rearrangements in B and T cells, respectively, and, based on the characteristics of the chromosomal breakpoint, can be broadly divided into three groups: (1) translocations derived from mistakes of the recombination activating gene RAG-mediated V(D)J recombination process, as is the case for translocations involving IGH and CCND1 in MCL or IGH and BCL2 in follicular lymphoma (FL)34–36; (2) translocations mediated by errors in the AID dependent CSR process, such as those involving the Ig genes and MYC in sporadic Burkitt lymphoma (BL)34; (3) translocations occurring as by-products of the AID-mediated SHM mechanism, which also generates DNA breaks, such as those joining the Ig and MYC loci in endemic BL.34 Conclusive experimental evidence for the involvement of antibody-associated remodeling events has been provided through in vivo studies performed in lymphoma-prone mouse models, where the removal of the AID enzyme was sufficient to abrogate the generation of MYC-IGH translocations in normal B cells undergoing CSR37,38 and to prevent the development of GC-derived B-NHL.39
The common feature of all NHL-associated chromosomal translocations is the presence of a proto-oncogene in the proximity of the chromosomal recombination sites. In most lymphoma types, and in contrast with acute leukemias, the coding domain of the oncogene is not affected by the translocation, but its pattern of expression is altered as a consequence of the juxtaposition of heterologous regulatory sequences derived from the partner chromosome (proto-oncogene deregulation) (Fig. 24.3). This process of proto-oncogene deregulation is defined as homotopic if a proto-oncogene whose expression is tightly regulated in the normal tumor counterpart becomes constitutively expressed in the lymphoma cell, and heterotopic when the proto-oncogene is not expressed in the putative normal counterpart of the tumor cell and undergoes ectopic expression in the lymphoma. In most types of NHL-associated translocations, the heterologous regulatory sequences responsible for proto-oncogene deregulation are derived from antigen receptor loci, which are expressed at high levels in the target tissue.34 However, in certain translocations, such as the ones involving BCL6 in diffuse large B-cell lymphoma (DLBCL), different promoter regions from distinct chromosomal sites can be found juxtaposed to the proto-oncogene in individual tumor cases, a concept known as promiscuous translocations.40–47
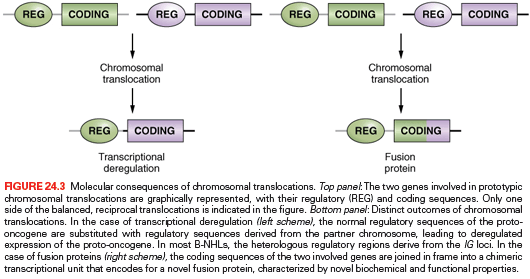
Less commonly, B-NHL associated chromosomal translocations juxtapose the coding regions of the two involved genes to form a chimeric unit that encodes for a novel fusion protein, an outcome typically observed in chromosomal translocation associated with acute leukemia (see Fig. 24.3). Examples are the t(11;18) of mucosa-associated lymphoid tissue (MALT) lymphoma and the t(2;5) of anaplastic large-cell lymphoma (ALCL). The molecular cloning of the genetic loci involved in most recurrent translocations has led to the identification of a number of proto-oncogenes involved in lymphomagenesis.
Aberrant Somatic Hypermutation
The term aberrant somatic hypermutation defines a mechanism of genetic lesion that appears to derive from a malfunction in the physiologic SHM process, leading to the mutation of multiple non-Ig genes.48 This phenomenon is uniquely associated with B-NHL and particularly with DLBCL, where over 10% of actively transcribed genes have been found mutated as a consequence of ASHM.
In GC B cells, SHM is tightly regulated both spatially and temporally to introduce mutations only in the rearranged IgV genes49 as well as in the 5′ region of a few other genes, including BCL6 and the CD79 components of the B-cell receptor,50–53 although the functional role of mutations found in these other genes remains obscure. On the contrary, multiple mutational events were found to affect numerous loci have been found mutated in over half of DLBCL cases and, at lower frequencies, in few other lymphoma types.54–58 The identified target loci include several well known proto-oncogenes such as PIM1, PAX5, and MYC, one of the most frequently altered human oncogenes.48 These mutations are typically distributed within ~2 kb from the transcription initiation site (i.e., the hypermutable domain in the Ig locus)59 and, depending on the genomic configuration of the target gene, may affect nontranslated as well as coding regions, thus holding the potential of altering the response to factors that normally regulate their expression or changing key structural and functional properties.48 This is the case of MYC, where a significant number of amino acid substitutions have proven functional consequences in activating its oncogenic potential. Nonetheless, a comprehensive characterization of the potentially extensive genetic damage caused by ASHM is still lacking, and the mechanism involved in this malfunction has not been elucidated.
Copy Number Gains and Amplifications
In addition to chromosomal translocations and ASHM, the structure of proto-oncogenes and their pattern of expression can be altered by CN gains and amplifications, leading to overexpression of an intact protein. Compared to epithelial cancer, only a few genes have been identified so far as specific targets of amplification in B-NHL, as exemplified by REL and BCL2 in DLBCL60–63 and by the genes encoding for programmed cell death 1 PD-1 ligands in primary mediastinal B-cell lymphoma (PMBCL).64,65
Activating Point Mutations
Somatic point mutations in the coding sequence of a target proto-oncogene may alter the biologic properties of its protein product, leading to its stabilization or constitutive activation. Over the past few years, the use of genomewide, high throughput sequencing technologies has allowed for the identification of numerous previously unsuspected targets of somatic mutations in cancer, including lymphoid malignancies. These genes will be discussed in individual disease sections. Of note, mutations of the RAS genes, a very frequent proto-oncogene alteration in human neoplasia, are rare in lymphomas.66
Inactivating Mutations and Deletions
Until recently, the TP53 gene, possibly the most common target of genetic alteration in human cancer,67 remained one of few bona fide tumor suppressor genes involved in the pathogenesis of NHL, although at generally low frequencies and restricted to specific disease subtypes, such as BL and DLBCL derived from the transformation of FL or CLL.68,69 The mechanism of TP53 inactivation in NHL is analogous to the one observed in human neoplasia in general, entailing a point mutation of one allele and a chromosomal deletion or mutation of the second allele. However, recent efforts taking advantage of genomewide technologies revealed several additional candidate tumor suppressor genes that are lost in B-NHL through specific chromosomal deletions and/or deleterious mutations. Two such genes lie on the long arm of chromosome 6 (6q), a region long known to be deleted in a large percentage of aggressive lymphomas associated with poor prognoses.70,71 The PRDM1/BLIMP1 gene on 6q21 is biallelically inactivated in ~25% of ABC-DLBCL cases,72–74 and the gene encoding for the negative nuclear factor kappa B (NF-κB) regulator A20 on chromosome 6q23 is commonly lost in ABC-DLBCL, PMBCL, and subtypes of marginal zone lymphoma and HL.75–78 Monoallelic inactivating mutations and deletions were found to affect the acetyltransferase genes CREBBP and EP300 in a significant proportion of DLBCL and FL, suggesting a role as haploinsufficient tumor suppressors.79 These two lymphoma types also harbor truncating mutations of MLL2,80 which is emerging as one of the most commonly mutated genes in cancer. Among the tumor suppressors preferentially inactivated by CN losses, it is important to mention the DLEU2/miR15-a/16.1 cluster on chromosome 13q14.3, the most frequent alteration in CLL (>50% of cases),81 and CDKN2A/B (p16/INK4a), which is inactivated by focal homozygous deletions in transformed FL (tFL), Richter syndrome (RS), and ABC-DLBCL,82–84 and less frequently, by epigenetic transcriptional silencing in various B-NHL.85
Infectious Agents
Viral and bacterial infections have both been implicated in the pathogenesis of lymphoma. At least three viruses are associated with specific NHL subtypes: the Epstein-Barr virus (EBV), the human herpesvirus-8/Kaposi sarcoma–associated herpesvirus (HHV-8/KSHV), and the human T-lymphotropic virus type 1 (HTLV-1). Other infectious agents, including HIV, hepatitis C virus (HCV), Helicobacter pylori, and Chlamydophila psittaci have an indirect role in NHL pathogenesis by either impairing the immune system and/or providing chronic antigenic stimulation.
EBV was initially identified in cases of endemic African BL,86,87 and was subsequently also detected in a fraction of sporadic BL, HIV-related lymphomas, and primary effusion lymphomas (PEL).88–92 Upon infection, the EBV genome is transported into the nucleus of the B lymphocyte, where it exists predominantly as an extrachromosomal circular molecule (episome).93 The formation of circular episomes is mediated by the cohesive terminal repeats, which are represented by a variable number of tandem repeats (VNTR) sequence.93,94 Because of this termini heterogeneity, the number of VNTR sequences enclosed in newly formed episomes may differ considerably, thus representing a clonal marker of a single infected cell.94 Evidence for a pathogenetic role of the virus in NHL infected by EBV is at least twofold. First, it is well recognized that EBV is able to significantly alter the growth of B cells.93 Secondly, EBV-infected lymphomas usually display a single form of fused EBV termini, suggesting that the lymphoma cell population represents the clonally expanded progeny of a single infected cell.88,89 Nonetheless, the role of EBV in lymphomagenesis is still unclear, because the virus infects virtually all humans during their lifetime and its transforming genes are commonly not expressed in the tumor cells of BL.
HHV-8 is a gammaherpesvirus initially identified in tissues of HIV-related Kaposi sarcoma95 and subsequently found to infect PEL cells as well as a substantial fraction of multicentric Castleman disease.96–98 Phylogenetic analysis has shown that the closest relative of HHV-8 is herpesvirus saimiri (HVS), a gamma-2 herpesvirus of primates associated with T-cell lymphoproliferative disorders.99 Like other gammaherpesviruses, HHV-8 is also lymphotropic, and infect lymphocytes both in vitro and in vivo.95,97,98 Lymphoma cells naturally infected by HHV-8 harbor the viral genome in its episomal configuration and display a marked restriction of viral gene expression, suggesting a pattern of latent infection.99
The HTLV-1 RNA retrovirus was first isolated from a cell line established from an adult T-cell leukemia/lymphoma (ATLL) patient.100 Unlike acutely transforming retroviruses, the HTLV-1 genome does not encode a viral oncogene nor does it transform T cells by cis-activation of an adjacent cellular proto-oncogene, because the provirus appears to integrate randomly within the host genome.101–103 The pathogenetic effect of HTLV-1 was initially attributed to the viral production of a trans-regulatory protein (HTLV-1 tax) that can activate the transcription of several host genes.104–110 However, Tax expression is suppressed in vivo, most likely to allow for an immune escape of the infected cells, questioning its role in transformation. More recently, a viral factor has been identified, which is thought to be involved in cell proliferation and viral replication and which may be responsible for HTLV-1–mediated lymphomagenesis.
An association between B-NHL and infection by HCV, a single stranded RNA virus of the Flaviviridae family, has been proposed based on the increased risk of developing lymphoproliferative disorders observed among HCV-positive patients111 and also on the results of interventional studies demonstrating that eradication of HCV with antiviral treatment could directly induce lymphoma regression in seropositive patients affected by indolent NHL.112 Although the underlying mechanisms remain unclear, current models suggest that chronic B-cell stimulation by antigens associated with HCV infection may induce nonmalignant B-cell expansion, which subsequently evolves into B-NHL by accumulating additional genetic lesions.
The causative link between antigen stimulation by H. pylori and MALT lymphoma originating in the stomach is documented by the observation that H. pylori can be found in the vast majority of the lymphoma specimens,113–115 and eradication of infection with antibiotics leads to long-term complete regression in 70% of cases.116 However, cases with t(11;18)(q21;21) respond poorly to antibiotic eradication,117 suggesting additional players.
C. psittaci, an obligate intracellular bacterium, was recently linked to the development of ocular adnexal marginal zone B-cell lymphoma (MZL), although variations in prevalence among different geographical areas remain a major investigational issue.118,119 In this indolent lymphoma, C. psittaci causes both local and systemic persistent infection, and presumably contributes to lymphomagenesis through its mitogenic activity and its ability to promote polyclonal cell proliferation and resistance to apoptosis in the infected cells in vivo. Notably, bacterial eradication with antibiotic therapy is often followed by lymphoma regression.120
MOLECULAR PATHOGENESIS OF B-NHL
The following section will focus on well-characterized genetic lesions that are associated with the most common types of B-NHL, classified according to the World Health Organization (WHO) classification of lymphoid neoplasia.1 The molecular pathogenesis of HIV-related NHL will also be addressed, whereas the pathogenesis of other B-cell NHLs remains far less understood. Lymphoblastic lymphoma, which is considered the same disease as T- and B-acute lymphoblastic leukemia, will not be covered in this chapter.
Mantle Cell Lymphoma
Cell of Origin
Mantle cell lymphoma is an aggressive disease representing ~5% of all NHL diagnoses and generally regarded as incurable.1 Based on immunophenotype, gene expression profile, and molecular features, such as the presence of unmutated IgV genes in the vast majority of cases, MCL has been historically considered as derived from naïve, pre-GC peripheral B cells located in the inner mantle zone of secondary follicles (see Fig. 24.1).121 More recently, the observation of BCR diversity, including IGHV hypermutation, in a subset of tumor cases (15% to 40%) has shifted this paradigm, suggesting the existence of distinct molecular subtypes, including one influenced by the CG environment.
Genetic Lesions
MCL is typically associated with the t(11;14)(q13;q32) translocation, that juxtaposes the IGH gene at 14q32 to a region containing the CCND1 gene (also known as BCL1) on chromosome 11q13.122–124 The translocation consistently leads to homotopic deregulation and overexpression of cyclin D1, a member of the D-type G1 cyclins that regulates the early phases of the cell cycle and is normally not expressed in resting B cells.125–127 By deregulating cyclin D1, t(11;14) is thought to contribute to malignant transformation by perturbing the G1-S phase transition of the cell cycle.121 Importantly, the frequency and specificity of this genetic lesion, together with the expression of cyclin D1 in the tumor cells, provides an excellent marker for MCL diagnosis.1
In addition to t(11;14), up to 10% of MCLs overexpress aberrant or shorter cyclin D1 transcripts, as a consequence of secondary rearrangements, microdeletions, or point mutations in the gene 3′ untranslated region.128–130 These alterations lead to cyclin D1 overexpression through the removal of destabilizing sequences and the consequent increase in the mRNA half-life, and are more commonly observed in cases characterized by high proliferative activity and a more aggressive clinical course. The pathogenic role of cyclin D1 deregulation in human neoplasia is suggested by the ability of the overexpressed protein to transform cells in vitro and to promote B-cell lymphomagenesis in transgenic mice, although only when combined to other oncogenic events131,132; however, an animal model that faithfully recapitulates the features of the human MCL is still lacking.
Other genetic alterations involved in MCL include biallelic inactivation of the ATM gene by genomic deletions and mutations,133 loss of TP53 (20% of patients, where it represents a marker of poor prognosis),134 and inactivation of the CDKN2A gene by deletions, point mutations, or promoter hypermethylation (approximately half of the cases belonging to the MCL variant characterized by a blastoid cell morphology).135 Also associated with aggressive tumors are mutations activating the Notch signaling pathway, including NOTCH1 (12% of clinical samples) and NOTCH2 (5% of samples); these lesions, which are mutually exclusive, mostly consist of truncating events that remove the PEST sequences required for NOTCH protein degradation and, thus, lead to protein stabilization.136,137 Other recurrent mutations were reported in genes encoding the antiapoptotic protein BIRC3, the Toll-like receptor 2 (TLR2), the chromatin modifiers WHSC1 and MLL2, and the MEF2B transcription factor.136 In a small number of cases, BMI1 is amplified and/or overexpressed, possibly as an alternative mechanism to the loss of CDKN2A.138,139
Burkitt Lymphoma
Cell of Origin
BL is an aggressive lymphoma comprising three clinical variants, namely sporadic BL (sBL), endemic BL (eBL), and HIV-associated BL, often diagnosed as the initial manifestation of AIDS.1 In all variants, the presence of highly mutated IgV sequences140–143 and the expression of a distinct transcriptional signature144,145 unequivocally confirm the derivation from a GC B cell.
Genetic Lesions
All BL cases, including the leukemic variants, share a virtually obligatory genetic lesion (i.e., chromosomal translocations involving the MYC gene on region 8q24 and one of the Ig loci on the partner chromosome).146,147 In ~80% of cases, this is represented by the IGH locus, leading to t(8;14)(q24;q32), whereas in the remaining 20% of cases, either IGκ (2p12) or IGλ(22q11) are involved.146–149 Although fairly homogeneous at the microscopic level, these translocations display a high degree of molecular heterogeneity, the breakpoints being located 5′ and centromeric to MYC in t(8;14), but mapping 3′ to MYC in t(2;8) and t(8;22).146–150 Further molecular heterogeneity derives from the exact breakpoint sites observed on chromosomes 8 and 14 in t(8;14): Translocations of eBL tend to involve sequences at an undefined distance (>1,000 kb) 5′ to MYC on chromosome 8 and sequences within or in proximity to the IGHJ region on chromosome 14.151,152 In sBL, t(8;14) preferentially involves sequences within or immediately 5′ to MYC (<3 kb) on chromosome 8 and within the IGH switch regions on chromosome 14.151,152
The common consequence of t(8;14), t(2;8) and t(8;22) is the ectopic and constitutive overexpression of the MYC proto-oncogene,153–155 which is normally absent in the majority of proliferating GC B cells,13 in part due to BCL6-mediated transcriptional repression.156 Oncogenic activation of MYC in BL is mediated by at least three distinct mechanisms: (1) juxtaposition of the MYC coding sequences to heterologous enhancers derived from the Ig loci;153–155 (2) structural alterations of the gene 5′ regulatory sequences, which affect the responsiveness to cellular factors controlling its expression157—in particular, the MYC exon 1/intron 1 junction encompasses critical regulatory elements that are either decapitated by the translocation or mutated in the translocated alleles; and (3) amino acid substitutions within the gene exon 2, encoding for the protein transactivation domain.158,159 These mutations can abolish the ability of p107, a nuclear protein related to RB1, to suppress MYC activity160 or increase protein stability.161,162
MYC is a nuclear phosphoprotein that functions as a sequence-specific DNA-binding transcriptional regulator controlling proliferation, cell growth, differentiation, and apoptosis, all of which are implicated in carcinogenesis.163 In addition, MYC controls DNA replication independent of its transcriptional activity, a property that may promote genomic instability by inducing replication stress.164 Consistent with its involvement in multiple cellular processes, the MYC target gene network is estimated to include ~15% of all protein-coding genes as well as noncoding RNAs.163,165 In vivo, MYC is found mainly in heterodimeric complexes with the related protein MAX, and such interaction is required for MYC-induced stimulation of transcription and cell proliferation.166–172 In NHL carrying MYC translocations, constitutive expression of MYC induces the transcription of target genes with diverse roles in regulating cell growth by affecting DNA replication, energy metabolism, protein synthesis, and telomere elongation.163,172,173 Furthermore, deregulated MYC expression is thought to cause genomic instability, and thus contributes to tumor progression by facilitating the occurrence of additional genetic lesions.174 Dysregulation of MYC expression in a number of transgenic mouse models leads to the development of aggressive B-cell lymphomas with high penetrance and short latency.162,175,176 These mouse models confirm the pathogenetic role of deregulated MYC in B cells, although the resulting tumors tend to be more immature than the human BL, most likely due to the early activation of the promoter sequences used for the expression of the MYC transgene.
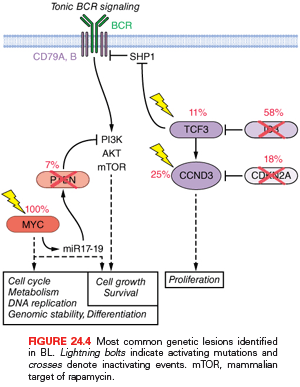
More recently, the application of new genomics technologies revealed additional oncogenic mechanisms that cooperate with MYC in the development of this aggressive lymphoma. Mutations of the transcription factor 3 (TCF3) (10% to 25%) and its negative regulator ID3 (35% to 58%) are highly recurrent in all three subtypes of BL, where they promote tonic (antigen-independent) BCR signaling and sustain survival of the tumor cell by engaging the phosphoinositide 3-kinase (PI3K) pathway (Fig. 24.4).177 In addition, TCF3 can promote cell-cycle progression by transactivating CCND3. Notably, CCND3 is itself a target of gain-of-function mutations in 38% of sBL, where these events affect conserved residues in the carboxyl terminus of this D-type cyclin, which are implicated in the control of protein stability, leading to higher expression levels. Interestingly, CCND3 mutations occur in only 2.6% of eBL, suggesting alternative oncogenic mechanisms in this subtype.177 Other common genetic lesions include the loss of TP53 by mutation and/or deletion (35% of both sBL and eBL cases),68 inactivation of CDKN2B by deletion or hypermethylation (17% of samples),85 and deletions of 6q, detected in ~30% of cases, independent of the clinical variant.70 Finally, one contributing factor to the development of BL is monoclonal EBV infection, present in virtually all cases of eBL and in ~30% of sBL.86,88,178,179 The consistent expression of EBER, a class of small RNA molecules, has been proposed to mediate the transforming potential of EBV in BL.180 However, EBV infection in BL displays a peculiar latent infection phenotype characterized by negativity of both EBV-transforming antigens LMP1 and EBNA2; thus, the precise pathogenetic role of this virus has remained elusive.181
Follicular Lymphoma
FL represents the second most common type of B-NHL (~20% of diagnoses) and the most common low-grade B-NHL.1 It is an indolent but largely incurable disease, characterized by a continuous pattern of progression and relapses that often culminates in its histologic transformation to an aggressive lymphoma with a diffuse large cell architecture and a dismal prognosis (20% to 30% of cases).182,183
Cell of Origin
The ontogeny of FL from a GC B cell is supported by the expression of specific GC B-cell markers such as BCL6 and CD10, together with the presence of somatically mutated Ig genes showing evidence of ongoing SHM activity.1
Genetic Lesions
The genetic hallmark of FL is represented by chromosomal translocations affecting the BCL2 gene on chromosome band 18q21, which are detected in 80% to 90% of cases independent of cytologic subtype, although less frequent in grade 3 FL.184–187 These rearrangements join the 3′ untranslated region of BCL2 to an IG JH segment, resulting in the ectopic expression of BCL2 in GC B cells,184,185,188–192 where its transcription is normally repressed by BCL6.18,193 Approximately 70% of the breakpoints on chromosome 18 cluster within the major breakpoint region, whereas the remaining 5% to 25% map to the more distant minor cluster region, located ~20 kb downstream of the BCL2 gene.184,185,188,189 Rearrangements involving the 5′ flanking region of BCL2 have been described in a minority of cases.194 The BCL2 gene encodes a 26-kd integral membrane protein that controls the cell apoptotic threshold by preventing programmed cell death and, thus, may contribute to lymphomagenesis by inducing apoptosis resistance in tumor cells independent of antigen selection. Nevertheless, additional genetic aberrations are required for malignant transformation. Most prominent among them are mutations in multiple epigenetic modifiers, including the methyltransferase MLL2 (80% to 90% of cases),80 the polycomb-group oncogene EZH2 (7% of patients),195 the acetyltransferases CREBBP and EP300 (40% of cases),79,80 and multiple core histones,196 which may all contribute to transformation by remodeling the epigenetic landscape of the precursor tumor cell. A major role is also played by chronic antigen stimulation.197,198
Whole-exome sequencing and CN analysis of sequential, clonally related FL and tFL biopsies has recently provided the ability to characterize the molecular events that are specifically acquired during histologic progression to DLBCL, and thus presumably play a major role in conferring this more aggressive phenotype. tFL-specific lesions include inactivation of CDKN2A/B through deletion, mutation, and hypermethylation (one-third of patients),84,136,199 rearrangements and amplifications of MYC,84,200 TP53 mutations/deletions (25% to 30% of cases),69,201–203 loss of chromosome 6 (20%),70 ASHM, and although larger cohorts of patients will need to be studied, biallelic loss of the immune regulator B2M.84 Chromosomal translocations of the BCL6 gene are detected in 6% to 14% of all FL cases, and were shown to have a significantly higher prevalence in the group of patients that undergo transformation into aggressive DLBCL.204–207
Diffuse Large B-Cell Lymphoma
DLBCL is the most common form of B-NHL, accounting for ~40% of all new diagnoses in adulthood and including cases that arise de novo, as well as cases that derive from the clinical evolution of various, less aggressive B-NHL types (i.e., FL and CLL).1,208
Cell of Origin
Based on gene expression profile analysis, at least three well-characterized molecular subtypes have been recognized within this diagnostic entity, which reflect the derivation from B cells at various developmental stages. Germinal center B-cell–like (GCB) DLBCL appears to derive from proliferating GC cells; ABC-DLBCL shows a transcriptional signature related to BCR-activated B cells or to B cells committed to plasmablastic differentiation; and PMBCL is postulated to arise from post-GC thymic B cells. The remaining 15% to 30% of cases remain unclassified.209–212 Stratification according to gene expression profiles has prognostic value, because patients diagnosed with GCB-DLBCL display better overall survival compared to ABC-DLBCL,63 but is imperfectly replicated by immunophenotyping or morphology and does not presently inform differential therapy213,214; thus, it is not officially incorporated into the WHO classification. A separate classification schema identified three discrete subsets defined by the expression of genes involved in oxidative phosphorylation, B-cell receptor/proliferation, and tumor microenvironment/host inflammatory response.215
Genetic Lesions
The heterogeneity of DLBCL is reflected in the catalog of genetic lesions that are associated with its pathogenesis, and include balanced reciprocal translocations, gene amplifications, chromosomal deletions, single point mutations, and relatively unique among all NHL, ASHM. During the past few years, the application of genomewide approaches such as whole-exome/transcriptome/genome sequencing and single nucleotide polymorphism (SNP) array analysis have provided a comprehensive picture of the DLBCL genomic landscape. One important finding of these studies is that, compared to other B-cell malignancies, DLBCL shows a significantly higher degree of genomic complexity, harboring on average 50 to more than 100 lesions per case, with great diversity across patients.80,216,217 Although many of the identified lesions can be variably found in both molecular subtypes of the disease, consistent with a general role during transformation, others appear to be preferentially or exclusively associated with individual DLBCL subtypes, indicating that GCB and ABC-DLBCL utilize distinct oncogenic pathways (Fig. 24.5).
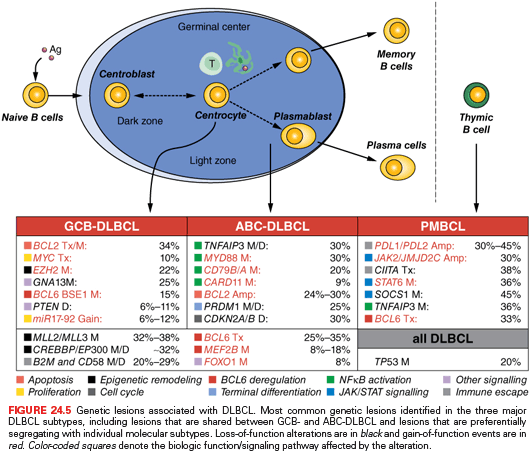
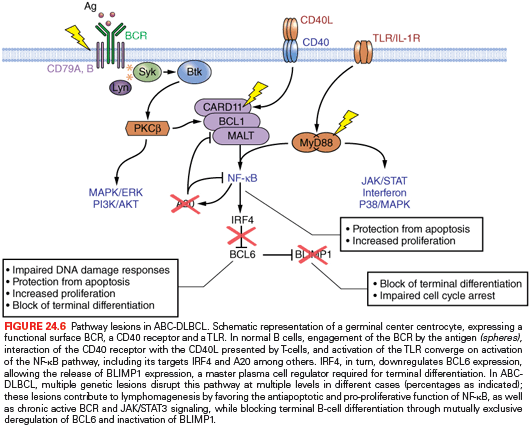
GCB and ABC Shared Lesions. The most prominent program disrupted in DLBCL, independent of subtype, is represented by epigenetic regulation of chromatin due to mutations in the CREBBP/EP300 acetyltransferase genes (35% of cases) and the MLL2 H3K4 trimethyltransferase (~30% of cases).79,80,217 These lesions may favor tumor development by reprogramming the cancer epigenome and, in the case of CREBBP/EP300, by altering the balance between the activity of the BCL6 oncogene, which is typically inactivated by acetylation, and the tumor suppressor p53, which requires acetylation at specific residues for its function.79
Deregulated activity of the BCL6 oncoprotein due to a multitude of genetic lesions is also a major contributor to DLBCL pathogenesis, in both GCB and ABC-DLBCL. Chromosomal rearrangements of the BCL6 gene at band 3q27 are observed in up to 35% of cases,71,205,218 although with a twofold higher frequency in the ABC-DLBCL subtype (see Fig. 24.4).219 These rearrangements juxtapose the intact coding domain of BCL6 downstream and in the same transcriptional orientation to heterologous sequences derived from the partner chromosome, including IGH (14q23), IGκ (2p12), IGλ (22q11), and at least 20 other chromosomal sites unrelated to the IG loci.40–47 The majority of these translocations result in a fusion transcript in which the promoter region and the first noncoding exon of BCL6 are replaced by sequences derived from the partner gene.41,220 Because the common denominator of these promoters is a broader spectrum of activity throughout B-cell development, including expression in the post-GC differentiation stage, the translocation is thought to prevent the downregulation of BCL6 expression that is normally associated with differentiation into post-GC cells. Deregulated expression of an intact BCL6 gene product is also sustained by a variety of indirect mechanisms, including gain-of-function mutations in its positive regulator MEF2B (~11% of cases),221 inactivating mutations/deletions of CREBBP/EP300,79 and mutations/deletions of FBXO11 (~5%),222 which encodes a ubiquitin ligase involved in the control of BCL6 protein degradation. These lesions play a critical role in lymphomagenesis by enforcing the proliferative phenotype typical of GC cells, while suppressing proper DNA damage responses; moreover, constitutive expression of BCL6 blocks terminal differentiation, as confirmed by a mouse model in which deregulated BCL6 expression causes DLBCL.223
DLBCL cells have also acquired the ability to escape both arms of immune surveillance, including cytotoxic T lympocytes (CTL)-mediated cytotoxicity (through genetic loss of the B2M/human leukocyte antigen class I [HLA-I] genes) and natural killer (NK) cell–mediated death (through genetic loss of the CD58 molecule).224 Analogous effects may be achieved in PMBCL by disruption of the major histocompatibility complex class II (MHC-II) transactivator class II major histocompatibility complex transactivator (CIITA) and amplification of the genes encoding for the immunomodulatory proteins PDL1/PDL2.
Finally, approximately 50% of all DLBCL are associated with ASHM.48 The number and identity of the genes that accumulate mutations in their coding and noncoding regions due to this mechanism varies in different cases and is still largely undefined. However, preferential targeting of individual genes has been observed in the two main DLBCL subtypes, with mutations of MYC and BCL2 being found at significantly higher frequencies in GCB-DLBCL, and mutations of PIM1 almost exclusively observed in ABC-DLBCL. ASHM may, therefore, contribute to the heterogeneity of DLBCL via the alteration of different cellular pathways in different cases. Mutations and deletions of the TP53 tumor suppressor gene are detectable in ~20% of cases, including those that originate from the transformation of FL, and are more often associated with chromosomal translocations involving BCL2.69
GCB-DLBCL. Genetic lesions specific to GCB-DLBCL include the t(14;18) and t(8;14) translocations, which deregulate the BCL2 and MYC oncogenes in 34% and 10% of cases, respectively.63,193,225,226 Also exquisitely restricted to this subtype are mutations of the EZH2 gene,195 which encodes a histone methyltransferase responsible for trimethylating Lys27 of histone H3 (H3K27), mutations of the S1PR2 adaptor protein GNA13, mutations affecting an autoregulatory domain within the BCL6 5′ untranslated exon 1,219,227,228 and deletions of the tumor suppressor PTEN.83,229
Somatic mutations of the BCL6 5′ regulatory sequences are detected in up to 75% of DLBCL cases52,230,231 and reflect the activity of the physiologic SHM mechanism that operates in normal GC B cells.52,53 However, a functional analysis of numerous mutated BCL6 alleles uncovered a subset of mutations that are specifically associated with GCB-DLBCL and that are not observed in normal GC cells or in other B-cell malignancies.227 These mutations deregulate BCL6 transcription by disrupting an autoregulatory circuit through which the BCL6 protein controls its own expression levels via binding to the promoter region of the gene227,228 or by preventing CD40-induced BCL6 downregulation in post-GC B cells.232 Because the full extent of mutations deregulating BCL6 expression has not been characterized, the fraction of DLBCL cases carrying abnormalities in BCL6 cannot be determined.
ABC-DLBCL. Several genetic abnormalities are observed almost exclusively in ABC-DLBCL, including amplifications of the BCL2 locus on 18q24233,234; mutations within the NF-κB (CARD11, TNFAIP3/A20),75,235 B-cell receptor (CD79B),236 and TLR (MYD88)237 signaling pathways; inactivating mutations and deletions of BLIMP172–74; chromosomal translocations deregulating the BCL6 oncogene; and deletion or lack of expression of the p16 tumor suppressor. Mutations of the ATM gene have been reported in a small subset of cases.238
A predominant feature of ABC-DLBCL is the constitutive activation of the NF-κB signaling pathway, initially evidenced by the selective expression of a signature enriched in NF-κB target genes, and by the requirement of NF-κB for proliferation and survival of ABC-DLBCL cell lines. This phenotype is sustained by a variety of alterations affecting positive and negative regulators of NF-κB, as well as other adaptor molecules converging on activation of NF-κB, specifically in this disease subtype. In up to 30% of cases, the TNFAIP3 gene, encoding for the negative regulator A20, is biallelically inactivated by mutations and/or deletions, thus preventing termination of NF-κB responses.75,76 The tumor suppressor role of A20 was documented by the observation that reconstitution of A20 knockout cell lines with a wild-type protein induces apoptosis and blocks proliferation, in part due to suppression of NF-κB activity.75,76 In an additional ~10% of ABC-DLBCL, the CARD11 gene is targeted by oncogenic mutations clustering in the protein coiled-coil domain and enhancing its ability to transactivate NF-κB target genes.235 Less commonly, mutations were found in a variety of other genes encoding for NF-κB components, overall accounting for over half of all ABC-DLBCL75 and suggesting that yet unidentified lesions may be responsible for the NF-κB activity in the remaining fraction of cases.
In addition to constitutive NF-κB activity, ABC-DLBCLs display evidence of chronic active BCR signaling, which is associated with somatic mutations affecting the immunoreceptor tyrosine-based activation motif (ITAM) signaling modules of CD79B and CD79A in 10% of ABC-DLBCL biopsy samples, but rarely in other DLBCLs.236 Moreover, silencing several BCR proximal and distal subunits is toxic to ABC-DLBCL. These findings provided genetic evidence in support of the development of therapies targeting BCR signaling,236 and indeed, kinase inhibitors that interfere with this signaling pathway are emerging as a new treatment paradigm for ABC-DLBCL.
Approximately 30% of ABC-DLBCL patients harbor a recurrent change in the intracellular Toll/interleukin-1 receptor domain of the MYD88 adaptor molecule, which has the potential to activate NF-κB as well as JAK/STAT3 transcriptional responses.237 Although the relationship between MYD88 mutations and TLR signaling has not been studied, MYD88 was shown to be required for the survival of ABC-DLBCLs, indicating a pathogenic role for TLR in this disease type.
A second important program that is disrupted by genetic lesions in ABC-DLBCL includes terminal B-cell differentiation. In up to 25% of ABC-DLBCL cases, the PRDM1 gene is inactivated by biallelic truncating or missense mutations and/or genomic deletions, as well as by transcriptional repression through constitutively active, translocated BCL6 alleles.72–74 The PRDM1 gene encodes for a zinc finger transcriptional repressor that is expressed in a subset of GC B cells undergoing plasma cell differentiation and in all plasma cells,239,240 and is an essential requirement for terminal B-cell differentiation.241 Thus, BLIMP1 inactivation contributes to lymphomagenesis by blocking post-GC B-cell differentiation. Consistently, translocations deregulating the BCL6 gene are exceedingly rare in BLIMP1 mutated DLBCLs, suggesting that BCL6 deregulation and BLIMP1 inactivation represent alternative oncogenic mechanisms converging on the same pathway (Fig. 24.6).
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


