SUSCEPTIBILITY TO LUNG CANCER: GENETIC SUSCEPTIBILITY AND CARCINOGENS IN TOBACCO SMOKE
Tobacco use is the most important environmental factor associated with the development of lung cancer. Approximately 85% of lung cancer occurs in current or former smokers. Cigarette smoke contains more than 60 known carcinogens, 20 of which have been convincingly shown to cause lung tumors in laboratory animals or humans.1 Of these, polycyclic aromatic hydrocarbons, such as benzo(a)pyrene, tobacco-specific nitrosamines, such as 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), and aromatic amines, such as 4-aminobiphenyl, appear to have an important role in cancer causation. Nitrosamines such as NNK induce lung tumors in mice independent of the route of administration. Among the polycyclic aromatic hydrocarbons, benzo(a)pyrene is the most extensively tested and the first to be detected in tobacco smoke. Its role in cancer tumorigenesis is well described, and its diol epoxide metabolite has been implicated as a cause of mutations in the TP53 gene.2 One of the carcinogenic effects of tobacco smoke in the lung is the formation of DNA adducts, leading to errors in DNA replication and resulting in mutations. DNA adducts have been identified in the bronchial tissue of patients with lung cancer. In current smokers, adduct levels correlate with the amount of tobacco smoke exposure.3,4 In addition, in former smokers, the age at smoking initiation has been inversely associated with levels of DNA adducts, suggesting that the prevention of smoking in adolescence is of utmost importance in decreasing lung cancer risks.
Although tobacco use can account for the majority of lung cancers, most chronic smokers still do not develop lung cancer. Differences in inherent susceptibility may be related to variations in carcinogen metabolizing enzymes, DNA repair mechanisms, chromosome fragility, and other homeostatic mechanisms. Among genes for carcinogen-metabolizing enzymes, polymorphisms in the cytochrome P-450 genes, CYP1A1, CYP2D6, and CYP2E1, and in mu-class glutathione S-transferase (GSTM1) have received the most attention. In addition to inherent susceptibility to the carcinogenic effects of tobacco smoke, large genomewide association studies have identified lung cancer susceptibility loci at 15q25, 5p15, and 6p21.5,6 In particular, polymorphisms in and around nicotinic cholinergic receptors at chromosome 15q25 appear to correlate with mRNA and protein expression of these receptors as well as functional changes in the calcium ion channel of the A5 nicotine receptor; these differences confer susceptibility to smoking behaviors.7
Researchers are optimistic that molecular epidemiology will help identify individuals at the highest risk of developing lung cancer. Such information, in addition to the smoking history, will be of great value in new lung cancer screening trials and in chemoprevention trials to identify persons at the highest risk of developing lung cancer.
MOLECULAR CHANGES IN PRENEOPLASIA
Before lung cancer is clinically recognizable, a series of morphologically distinct changes (hyperplasia, metaplasia, dysplasia, and carcinoma in situ) are thought to occur. It is believed that dysplasia and carcinoma in situ represent true preneoplastic changes. It is evident that preneoplastic cells contain several genetic abnormalities identical to some of the abnormalities found in overt lung cancer cells. For squamous cell carcinomas (SQCC), an immunohistochemical analysis has confirmed the abnormal expression of proto-oncogenes (cyclin D1) and TSGs (p53).8 Allelotyping of precisely microdissected, preneoplastic foci of cells shows that 3p allele loss is currently the earliest known change, suggesting that one or more 3p TSGs may act as gatekeepers for lung cancer pathogenesis.9 This is followed by 9p, 8p, and 17p allele loss and p53 mutation. Similarly, atypical alveolar hyperplasia, the potential precursor lesion of adenocarcinomas, can harbor KRAS mutations and allele losses of 3p, 9p, and 17p.10 Other genetic alterations, such as inactivation of LKB1, whose germline mutations cause Peutz-Jeghers syndrome, have also been implicated in the development of adenocarcinoma. These observations are consistent with the multistep model of carcinogenesis and a field cancerization process, whereby the whole tissue region is repeatedly exposed to carcinogenic damage and is at risk for the development of multiple, separate foci of neoplasia.11 Although all types of lung cancers have associated molecular abnormalities in their normal and preneoplastic lung epithelium, SCLC patients in particular appear to have multiple genetic alterations occurring in their histologically normal-appearing respiratory epithelium.
Molecular changes have been found not only in the lungs of patients with lung cancer, but also in the lungs of current and former smokers without lung cancer. These molecular alterations are thus important targets for use in the early detection of lung cancer and for use as surrogate biomarkers in following the efficacy of lung cancer chemoprevention. The challenge is to identify not only the prevalence and temporal sequence of molecular lesions in lung preneoplasia, but also to determine which are rate limiting and indispensable and thus represent potential candidates for intermediate biomarker monitoring and therapeutic efforts.
GENETIC AND EPIGENETIC ALTERATIONS IN LUNG CANCERS
Genomic Instability and DNA Repair Genes
Similar to other epithelial tumors, lung cancer cells typically display chromosomal instability—both numeric abnormalities (aneuploidy) of chromosomes and structural cytogenetic abnormalities.12 Allele loss on chromosome 3p is thought to be the among the earliest genetic change occurring in both NSCLC and SCLC.13 In addition, nonreciprocal translocations and recurrent losses involving 1p, 4p, 4q, 5q, 6p 6q, 8p, 9p, 11p, 13q, 17p, 18q, and 22q may occur, representing changes in known and potential tumor suppressor genes (TSGs).14 Polysomes or regions of gene amplifications also occur and often involve proto-oncogenes, such as epidermal growth factor receptor (EGFR) and myelocytomatosis viral oncogene homolog (MYC).15,16 Simple reciprocal translocations are uncommonly observed in lung cancer, although translocations that give rise to BRD4-NUT, CRCT1-MAML2, SLC34A2–ROS1, and EML4-ALK fusion proteins have been recently reported.17–19 ALK and ROS1 translocations have been shown to drive the development and proliferation of tumors, and targeted therapy has resulted in clinical responses in patients with these abnormalities (see the following). In addition, alterations in microsatellite polymorphic repeat sequences are found in 35% of SCLC and 22% of NSCLC.20 Most recently, a comprehensive genomic analysis of lung squamous cell cancers has revealed a high-frequency selective amplification of chromosome 3q compared to lung adenocarcinomas.21 Furthermore, large-scale genomic profiling has established that there is a higher rate of copy number alterations, somatic rearrangements, and mutations found in squamous and adenocarcinoma lung cancers compared to other tumors.22 For example, squamous cell lung cancers exhibit 8.1 somatic mutations per megabase (Mb) compared to other cancers where the frequency ranges from 1.0 to 3.2 mutations per megabase. These studies highlight the increased genetic complexity, genomic instability, higher mutation rate, and thus more extensive molecular heterogeneity that typifies lung cancer compared to other tumor types.
Recurring mutations in DNA repair genes are becoming increasingly recognized in multiple solid tumor types, including lung cancer. Ataxia telangiectasia mutated (ATM) plays a central role in the DNA damage response pathway that is triggered after genomic insults, including errors during replication, exposure to genotoxic drugs, or ionizing radiation, and has been identified in lung adenocarcinomas.22–24 Alterations in other DNA repair genes have been identified, including BRCA1 and BRCA2, proteins that are both involved in the repair process of homologous recombination especially for double-strand breaks, as well as ATR, which functions in sensing and activating DNA repair in response to single-stranded DNA breaks.22 Additionally, there is growing evidence that chromatin remodeling genes are highly mutated in lung cancer. Specifically, it has been recently shown that alterations in MLL2, MLL3, MLL4 (a group of histone-modifying genes) cluster in lung cancers, along with mutations in SMARCA4 and ARID1A (members of the SWI/SNF chromatin-remodeling complex).21–24 Ongoing studies will serve to further evaluate the role of these putative TSGs in lung tumorigenesis.
The most powerful tumor surveillance mechanism is involved in DNA damage response and repair of errors in DNA replication.25 The DNA glycosylase 8-oxo guanine (OGG1) specifically excises the oxidatively damaged mutagenic base 8-hydroxyguanine, which causes G:C→T:A transversions frequently found in lung cancer. Proof that OGG1 is central to this DNA repair is the evidence that lung adenocarcinoma spontaneously develops in OGG1 knockout mice and 8-hydroxyguanine accumulates in their genomes.26,27 Individuals with low OGG activity have a greatly increased risk of developing lung cancer. Polymorphisms in other DNA repair genes including ERCC1, XRCC1, ERCC5/XPG, and MGMT/AGT have been correlated with a reduction of polyaromatic hydrocarbon DNA adduct formation as well as with lower lung cancer risks in case-control studies. Furthermore, high expression of ERCC1 is associated with decreased response to platinum-based chemotherapy, and overexpression of ERCC1 correlates with better overall prognosis in NSCLC,28 reflecting improved repair of lethal DNA damage by platinum on the one hand, and greater DNA stability with less aggressive disease course on the other. More recent studies, however, have called into question the predictive ability of ERCC1 expression alone to predict cisplatin response, at least by immunohistochemical detection.29 Similarly, ribonucleotide reductase M1 (RRM1) overexpression correlates with better de novo prognosis but resistance to gemcitabine.30,31 High levels of the mismatch repair protein MSH2 have also been shown to be associated with poor survival with cisplatin, and may enhance the predictive significance of ERCC1 expression.32 Prospective clinical trials investigating ERCC1 and RRM1 biomarker-guided therapy are currently underway.
PROTOONCOGENES, GROWTH FACTOR SIGNALING, AND GROWTH FACTOR–TARGETED THERAPIES
Advances in the understanding of lung carcinogenesis have yielded to a rapid discovery of novel driver oncogenes activated by mutations, translocations, or gene amplification. These alterations occur in genes that encode receptors or intracellular proteins crucial for cellular growth, proliferation, and survival, driving tumor formation and maintenance.
Receptor Tyrosine Kinases
EGFR is one of the most commonly mutated proto-oncogenes in lung adenocarcinoma. It is a transmembrane receptor-tyrosine kinase that is normally activated by binding with one of its ligands, members of the EGF family. EGFR-sensitizing mutations lead to constitutive activation of the tyrosine kinase and phosphorylation of downstream pathways ultimately resulting in uncontrolled proliferation, invasion, and metastasis. In 2004, investigators first noted mutations within the tyrosine-kinase domain of EGFR in patients who had sustained dramatic responses to the EGFR tyrosine-kinase inhibitors (TKIs), erlotinib and gefitinib.33–35 The frequency of such mutations varies from approximately 10% of lung adenocarcinomas in North American and European populations to as high as 50% in Asia.36–39 The leucine to arginine substitution at position 858 (L858R) in exon 21 and short in-frame deletions in exon 19 are the most common sensitizing mutations, comprising approximately 90% of cases. Sensitizing EGFR mutations are both prognostic for longer survival irrespective of therapy and predictive of response to EGFR TKIs.35,39–47 Other mutations have been described within the EGFR gene, and for some, the responsiveness to EGFR TKI-directed therapy is similar to the deletion 19 and L858R, including the L861Q and the G719X mutations. An EGFR sequencing analysis of more than 1,000 patients with adenocarcinoma showed that 27 patients (2.5%) carried exon 20 insertions, representing approximately 9% of all EGFR mutations.48 These mutations are found in patients who share the same phenotype as the canonical sensitivity mutations within exon 19 and 21; however, treatment with EGFR TKIs does not result in dramatic responses, and the prognosis is more analogous to that of patients with wild-type EGFR. A major area of investigation has been the mechanisms of acquired resistance to EGFR TKIs. Both in vitro as well as rebiopsy studies from patients who have been treated and subsequently progressed on treatment have shown two broad resistance mechanisms, secondary EGFR alterations and non-EGFR bypass mechanisms.49 Approximately 50% of EGFR TKI resistance is due to a second site mutation, the T790M mutation occurring within exon 20.50,51 Other, non-EGFR mechanisms of TKI resistance include MET amplification, PIK3CA mutations, and change in histology to a small cell appearance.49 The T790M mutation is also found to be present at a very low prevalence in the germ line and appears to increase lung cancer risk independent of smoking.52
ERBB2 (formerly human epidermal growth factor receptor 2 [HER2]/neu) is a member of the erbB receptor family, along with EGFR. It is overexpressed in about 20% of NSCLCs, and amplified in 2% to 4% of cases.53 ERBB2 mutations have been identified in approximately 2% of NSCLCs, comprising in-frame insertions of 3 to 12 base pairs into exon 20, usually around the codon 776.34,38,54 A meta-analysis suggested that ERBB2 overexpression is an indicator of poor survival in lung adenocarcinoma,55 but amplification or mutations do not seem to be prognostic.55,56 Early trials examining targeted therapies selected patients on the basis of ERBB2 overexpression and were mostly unsuccessful.57 However, recent data suggest that trastuzumab, a recombinant humanized monoclonal antibody against ERBB2, may be active in ERBB2-mutant lung adenocarcinomas.34 In addition, irreversible TKIs targeting ERBB2 are in early clinical trials.
Hepatocyte growth factor receptor (HGFR) is a receptor tyrosine kinase encoded by MET. MET amplification has been reported in about 2% of NSCLCs, and has been implicated in up to 20% of cases with acquired resistance to EGFR TKIs.58 Several therapeutic strategies, including antibodies that target HGFR and anti-HGFR TKIs are under clinical evaluation in NSCLC patients, mostly aiming to prevent or overcome EGFR TKI resistance.59–61
Three other receptors have been nominated as potential druggable targets, especially in lung SQCC.62,63 Insulin-like growth factor receptor 1 (IGFR1) is a tyrosine kinase receptor that activates the MAPK and PI3K/AKT/mTOR pathways, playing an important role in cancer growth and progression.64 IGFR1 expression is higher in lung SQCC, and gene copy number has been associated with survival.65 Despite an initial promise, phase III studies of some anti-IGFR1 agents has halted due to excess toxicities, including treatment-related deaths in the experimental arm. Fibroblast growth factor receptor 1 (FGFR1) is a member of the family of fibroblast growth factor (FGF) receptors that are dysregulated in a variety of cancers. FGFR1 amplification has been described in approximately 20% of lung SQCCs and has been associated with cigarette smoking and worse survival.66,67 In vitro studies showed activity with anti-FGFR inhibitors in cell lines harboring this feature, and pan-FGFR and multikinase inhibitors are in early phases of clinical investigation. Discoidin death receptor 2 (DDR2) is a member of the DDR family of transmembrane receptors that signals through SRC and STAT pathways. Mutations in the kinase domain have been reported in nearly 4% of lung SQCCs and were shown to yield malignant transformation in vitro.68 Interestingly, some FDA-approved agents for chronic myelogenous leukemia seem to be active against the DDR family, including dasatinib, nilotinib, and imatinib.69
Translocated Genes in Lung Cancer
The anaplastic lymphoma kinase (ALK) fusion protein is an activating oncogenic driver of lung adenocarcinomas occurring in 3% to 7% of all NSCLCs.70,71 Multiple ALK variants have been identified in lung cancer, and different ALK partners like kinesin family member 5B (KIF5B) have been described.70,72,73 Similar to EGFR mutations, ALK fusions occur almost exclusively in adenocarcinomas, specifically with acinar histology, in never or former light smokers.70,74–76 ALK fusions are not found in tumors with mutations in EGFR or KRAS, and patients appear to be of younger age compared to those with EGFR mutant tumors.76 The gold standard test for ALK translocations is fluorescence in situ hybridization (FISH), but a variety of methods have been proposed, including immunohistochemistry, reverse transcription polymerase chain reaction (RT-PCR), and next-generation sequencing (NGS). The clinical activity of crizotinib—an oral inhibitor against c-MET, ALK, and ROS1—was impressive in early trials involving patients with the EML4-ALK translocation.71 In a randomized phase III trial, crizotinib significantly improved progression-free survival (PFS), response rate (RR), and quality of life in comparison to second-line chemotherapy among patients with metastatic NSCLC harboring ALK translocations.77 In a fashion analogous to that seen with EGFR TKI resistance, initial reports of resistance mechanisms include both ALK dependent (e.g., secondary mutations within ALK or amplification of the mutant ALK allele) and bypass tracks (e.g., the development of secondary KRAS and EGFR mutations have been described).78–81 Importantly, several second-generation inhibitors are under clinical investigation and have shown promising activity in patients with acquired resistance to crizotinib.82,83
More recently, fusions involving the receptor tyrosine kinases ROS1 and RET have been described in NSCLC, each with oncogenic activity and potential utility as clinical biomarkers. ROS1 belongs to the insulin receptor family and is not normally expressed in the human lung tissue.84 It has been demonstrated that ROS1 fusions induce autophosphorylation and downstream activation of common growth and survival pathways like mitogen-activated protein kinase (MAPK), STAT3, and PI3K/AKT. ROS1 rearrangements occur in approximately 1% to 2% of NSCLC patients, mostly in adenocarcinoma histology, younger patients, and never smokers.85 It has been shown that selective ALK inhibitors are also active in tumors harboring ROS1 translocations, both in vitro and in vivo.86 RET rearrangements were only described in NSCLC in 2011, involving KIF5B as a partner.87,88 Subsequently, CCD6, NCOA4, and TRIM33 have been described as alternative RET fusion partners.89,90 Similar to ALK translocations, RET partners contain a coiled-coil domain that functions as a dimerization unit, leading to ligand-independent homodimerization of the fused protein and constitutive activation of the RET kinase activity mediated by autophosphorylation. Like ALK and ROS1, RET fusions are associated with a lack of smoking history and adenocarcinoma histology, with a frequency slightly higher than 1% in all comers.91 Agents including cabozantinib,90 sorafenib, sunitinib, vandetanib, and ponatinib are being evaluated in this patient population.
Intracellular Signaling
Because downstream effectors are required for intracellular transduction of incoming growth factors or receptor signals, it is not surprising that proteins important in cytoplasmic signal transduction cascades are also implicated in carcinogenesis. The RAS gene family—in particular, KRAS—can be activated by point mutations at codons 12 or 13 in exon 2 in approximately 20% to 30% of NSCLCs. KRAS mutations more commonly occur in patients with significant smoking history and adenocarcinoma histology.92 Approximately 70% are G→T transversions, with the substitution of glycine by either cysteine or valine. Similar G→T transversions are observed in the mutated p53 gene, representing similar DNA damage as a result of bulky DNA adducts caused by the polycyclic hydrocarbons and nitrosamines in tobacco smoke. In a meta-analysis of 3,779 lung cancer patients, mutant KRAS was correlated to a worse prognosis in adenocarcinoma histology, but this link was not confirmed in a prospective study.93 The poor prognosis has been proposed to be related to the fact that KRAS mutations rarely occur with EGFR mutations, and EGFR mutant tumors have a better prognosis than EGFR wild type. Thus, if KRAS mutations are evaluated only in EGFR wild-type tumors, the adverse prognostic effect of KRAS disappears.94 Although the oncogenicity of aberrant KRAS is well established, effective agents that inhibit KRAS have not reached the clinic. Another strategy has been to target downstream effectors of RAS, like the RAF family proteins and MEK. For instance, sorafenib—a broad-spectrum kinase inhibitor including the RAF family—led to a disease control rate of 79% in the BATTLE trial95 among patients with either KRAS or BRAF mutations. However, these findings were not confirmed in a large phase III trial.96 The addition of selumetinib (AZD6244), a MEK1/2 inhibitor, to docetaxel significantly improved RR and PFS in a randomized, phase II trial.97 A phase III study evaluating this combination in patients with KRAS-mutant NSCLC is ongoing. Another MEK inhibitor, trametinib (GSK1120212), also showed promising results in combination with chemotherapy in early phase I/Ib studies.98,99 However, it is not clear whether these agents have a differential effectiveness according to the KRAS status, because similar responses have been observed in KRAS wild-type tumors.
The RAF proteins are serine/threonine kinases downstream of RAS, represented by three isoforms in mammals (ARAF, BRAF, and RAF1 [also known as CRAF]). BRAF is a known oncogene in lung adenocarcinoma, being mutated in approximately 3% of cases.100 In contrast to melanoma, where the majority of BRAF mutations occur at valine 600 (V600E) within exon 15, V600E comprises only half of cases in lung adenocarcinoma. Interestingly, BRAF mutations are more likely to occur in former/current smokers. Dabrafenib (BRF113928)—a specific V600E BRAF inhibitor—demonstrated a promising RR of 40% in a phase II trial involving patients with lung adenocarcinoma harboring this mutation.101 Dramatic responses have also been reported with vemurafenib, another specific inhibitor that is U.S. Food and Drug Administration (FDA)-approved for the treatment of melanoma.102 Apart from BRAF, recurrent mutations at ARAF and RAF1 inhibitory sites were recently described in lung adenocarcinoma. Importantly, these mutations seem to be oncogenic in vitro, and may predict a response to targeted therapies, including sorafenib and MEK inhibitors.103
MAP2K1 (also known as MEK1) is a serine/threonine kinase downstream of RAF. In NSCLC, point mutations have been identified in 1% of cases, comprising three major nucleotide substitutions (Q56P, K57N, and D67N).104 These mutations tend to be nonoverlapping with other drivers and induce constitutive extracellular signal-regulated kinase (ERK) phosphorylation and cell proliferation in vitro. It is still unknown if these mutations will predict clinical benefit from anti-MEK therapies.
Another frequently altered intracellular pathway in lung cancer is PI3K–AKT–mammalian target of rapamycin (mTOR). Phosphatidylinositol 3-kinases (PI3Ks) are lipid kinases that participate in regulating cell growth, proliferation, and survival. They comprise heterodimers formed by an 85-kDa regulatory subunit and a 110-kDa catalytic subunit, the latter being encoded by PIK3CA. PIK3CA mutations have been reported in several cancer types, including a frequency of 1% to 4% in lung adenocarcinoma.105,106 However, a higher frequency of 16% was reported in the lung squamous section of The Cancer Genome Atlas (TCGA),107 suggesting a predominance in this histologic subtype. The most common hotspots are at codons E542 and E545 in exon 9 (helical domain) and at codon H1047 in exon 20 (kinase domain). As opposed to other drivers, PIK3CA mutations often coexist with other oncogenic mutations in the case of lung adenocarcinoma and are not associated with smoking status.108 PIK3CA mutations were shown to induce constitutive pathway activation and cell transformation in vitro109 and have been associated with poor clinical prognosis.110 The best strategy to target PI3K and the population to be selected is still a matter of debate.
AKT1 is downstream of PI3K and is a member of a family of serine-threonine kinases. Recurrent mutations within the plekstrin homology/lipid binding domain of AKT1 at E17K have been described in up to 1% of patients with lung adenocarcinoma.111 This mutation causes constitutive activation of AKT and leads to cellular transformation in vitro and in vivo.112
Phosphatase and tensin homolog (PTEN) is a lipid/protein phosphatase that is integral to cellular processes and acts as a TSG gene by inhibiting the activity of the PI3K pathway. While germ-line mutations in PTEN lead to Cowden syndrome, somatic mutations have been found in patients with lung cancer, as well as epigenetic inactivation. PTEN mutations may be more common in smokers and those with squamous cancer, and are also seen in patients with SCLC.113
THE TECHNOLOGY OF GENOMIC ANALYSES OF LUNG CANCER
In recent years, technologic advances have enabled the comprehensive analyses of gene expression, copy number alterations, mutations, and other genetic perturbations across a large number of tumors.114 In the lung cancer setting, such global approaches have led to the understanding of cancer biology and the molecular mechanisms underlying key cancer phenotypes at an unprecedented scope. More importantly, this knowledge has leveraged the identification of subsets of tumors whose biology may be affected by interventions targeted against key dysregulated genes or pathways.115 These efforts have been increasingly applied in individual institutions or collaborative groups as will be described.
Although direct dideoxynucleotide (ddNTP) sequencing (or Sanger sequencing) has traditionally been the gold standard for the detection of recurrent point mutations in NSCLC (like EGFR), newer methodologies have been developed that are not only less dependent on the tumor fraction in the sample, but also simultaneously determine the mutational status of many genes in a single reaction. Multiplex genotyping comprises methods such as Sequenom (Sequenom, San Diego, CA) and SNaPShot (Applied Biosystems, Foster City, CA).116,117 In an early report using these techniques from the Massachusetts General Hospital in 2009, at least one alteration was evident in 51% of 552 cases, and no overlap was observed among the main driver oncogenes.118 More recently, the Lung Cancer Mutation Consortium (LCMC)—a collaborative group that genotyped 1,000 lung adenocarcinomas in the United States—detected genetic alterations in 54% of cases, and 97% were mutually exclusive.119
NGS platforms, also known as massively parallel sequencing, offer a wide range of opportunities to characterize the cancer genome for DNA, mRNA, transcription factor binding sites, microRNA (miRNA), and DNA methylation patterns.120,121 In TCGA, 230 lung adenocarcinomas and adjacent samples were analyzed using various NGS platforms.122 A whole-exome approach was used in all cases, whereas whole-genome sequencing was performed in 34 samples. The authors found a very high mutation rate (median 4.56 mutations per megabase), consistent with a median of 159 mutations per tumor (range, 13 to 1,339). As demonstrated in prior experiences, this was among the highest rates in TCGA projects, only behind lung SQCC and melanoma. Somatic mutations were found in known oncogenes, like KRAS, EGFR, and BRAF, as well as chromatin modifiers as ARID1A, SMARCA4, and SETD2. A number of recurrent noncanonical mutations were documented, including a MET Y1003* mutation. Lung SQCC was also explored in TCGA, including 178 pairs of tumor/normal samples from patients with stages I through IV disease.107 A total of 48,690 nonsilent mutations were identified through whole-exome sequencing, with a mean of 360 total exonic mutations per tumor (median, 8.4 per megabase). Ten genes were considered to harbor significant mutations, including TP53, CDKN2A, PTEN, PIK3CA, KEAP1, MLL2, HLA-A, NFE2L2, NOTCH1, and RB1. Frequent alterations were present in the following pathways: CDKN2A/RB1 (cell cycle control), NFE2L2/KEAP1/CUL3 (response to oxidative stress), PI3K/AKT (apoptotic signaling), and SOX2/TP63/NOTCH1 (squamous differentiation). In addition, whole-transcriptome sequencing and microarray expression was carried out. The mRNA expression data clustered in four distinct subtypes named classical, basal, secretory, and primitive, which correlate with miRNA, methylation, somatic copy number alterations, and mutation spectrum.
As a more comprehensive insight into the lung cancer genome emerges, it becomes clear that its complex biology will require continuous efforts and more integrative analyses. For instance, it is somewhat disappointing that approximately half of lung adenocarcinomas still lack a plausible target for drug development. It comes without saying that many alterations are detected in TSGs, for which no targeted therapies are available. These observations may also serve to argue that a fraction of driver mechanisms may be mediated by epigenetic or other regulatory mechanisms, as opposed to structural genomic alterations. One approach to fill this gap is the coordinated analysis of multiple types of data from large cohorts like TCGA. For example, integrating mutational data to gene expression, miRNA, or methylation profiling could potentially determine associations with particular sets of genes. Such gene sets could be subjected to computational analyses and biochemical studies to discover the biologic and clinical significance, as well as proposing alternative therapeutic strategies. The Clinical Proteomic Tumor Analysis Consortium (CPTAC) is an ongoing program that exemplifies the integration of high-throughput cancer genomics and proteomics using samples from TCGA, as well as cell lines and xenograft models.123
It is also intriguing that current genomic studies rely primarily on computational approaches to identify driver events and distinguish them from the background of passenger mutations. Such analyses may be flawed not only by the relatively low sample size, but also by the filtering method on its own.124,125 Indeed, most comprehensive genomic studies (as previously described) included a few more than a hundred samples and are not empowered to detect rare mutations. Larger efforts will be essential to achieve a larger number of cases per cancer type, as exemplified by TCGA and the International Cancer Genome Consortium. In addition, continuously improving the accuracy of analytical methods will be key to avoid false discoveries and to avoid overlooking potential targetable alterations.
EPIGENETICS/EPIGENOME
Over the last 20 years, it has become apparent that epigenetics dysregulation is nearly universal in cancers, and specifically in lung cancer. Indeed, a recent study showed that the determination of the methylation status of a four gene panel (CDKN2A, CDH13, RASSF1A, and APC) is predictive of lymph node recurrence in stage I cancers.126 A separate study confirmed the prognostic impact of a separate panel of methylated genes.127 Unfortunately, despite the near universal finding of hypermethylation of tumor-relevant genes and the prognostic impact of transcriptional silencing of these genes, attempts at interfering with this epigenetic program by using hypomethylating agents (usually in conjunction with histone deacetylase inhibitors) has failed to have a dramatic impact on typical tumor response criteria, although each of these trials showed tantalizing evidence of biologic effect.128–130
More recently, mutations within epigenetic modifying genes have been discovered in a variety of thoracic tumors. These include mutations within the SWI/SNF complex proteins SMARCA4/BRG1 and ARID1A.24 The SWI/SNF complex is an important chromatin modeling complex that is essential in yeast and is thought to function as a tumor suppressor in many human tumors, including lung cancers. Other mutations or overexpression of other epigenetic modifiers, including histone methyltransferases, histone acetyltransferase coactivators and the histone lysine demethylase KDM2A, have been described as well.21,131,132 Whether these mutations or overexpression of genes act as drivers of oncogenesis or act as passenger mutations/alterations has not yet been clarified. Furthermore, if these findings will serve as useful predictors of response to epigenetic therapy will depend on the development of well-designed prospective biomarker-driven and/or stratified trials.
TUMOR SUPPRESSOR GENES AND GROWTH SUPPRESSION
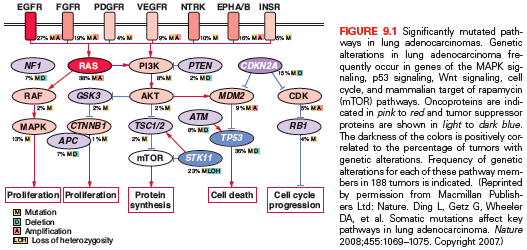
A number of TSGs have been identified that inhibit lung tumorigenesis or suppress key phenotypes in developed lung carcinomas. Many classical tumor suppressors have been identified by decades of work examining genes involved in human-inherited cancer syndromes and elucidating their role in sporadic cancers. Germ-line mutations in some TSGs, such as LKB1/STK11, p53, and RB1, give rise to inherited tumor syndromes; however, a somatic loss of TSGs within sporadic cancers is more commonly seen, including p53, PTEN, CDKN2A (encoding both p16INK4A and p14ARF), ATM, NF1, RB1, and APC. These have all been found to be mutated or lost in lung cancer through multiple mechanisms, such as inactivating mutations, chromosomal loss, methylation, or overexpression of other proteins that inhibit the suppressive gene’s expression or activity.23,24 We discuss several of the most frequently lost TSGs in the following paragraphs.
p53 Pathway
The p53 gene, located at chromosome 17p13, is crucial for maintaining genomic integrity in the face of cellular stress from DNA damage through gamma and ultraviolet irradiation, carcinogens, and chemotherapy. It is the most frequently mutated TSG in human malignancies, and mutations affect approximately 90% of SCLCs and >50% of NSCLCs. In NSCLCs, p53 alterations occur more frequently in SQCC (81%) than adenocarcinomas (~50%).21,22,24 p53 mutations have been linked to poorer prognosis retrospectively and in the TCGA Pan-Cancer analysis; however, they have not been shown to correlate with survival in a prospective randomized clinical trial.133,134 Most p53 mutations are G→T transversions, which correlate with cigarette smoking.2 The Li-Fraumeni syndrome of inherited germline p53 mutation may also lead to increased susceptibility to lung cancer in adults; this risk is magnified by tobacco smoking, because carriers who smoked had a 3.16-fold higher risk for lung cancer than nonsmokers.135 The majority of missense mutations occur in the DNA-binding domain of the protein, and five of the six most prevalently mutated sites are arginine residues that are involved with electrostatic interactions with DNA strands.136 Missense mutations prolong the half-life of the p53 protein, leading to increased protein levels detectable with immunohistochemistry. Also, because p53 exerts its cellular actions as a tetramer, mutant forms of the protein appear to exert dominant negative effects on wild-type p53 and have also been shown to inhibit the function of p63 and p73 family members.137
In addition to mutational or deletional loss of p53, other regulatory components of the p53 pathway are altered in lung cancer, including the ATM gene, the p53 binding protein MDM2, and the p14ARF tumor suppressor. ATM and the related protein ATR are tumor-suppressive serine/threonine kinases that activate cell cycle checkpoints in response to DNA damage and, ultimately, activate and stabilize p53.138 Although ATR and the downstream checkpoint kinases (CHEK) are mutated in less than 1% of lung adenocarcinomas, ATM has been found to have deleterious mutations in 7% of lung adenocarcinomas. These mutations were largely mutually exclusive with p53 mutations, likely indicating that mutations in both genes would have redundant effects, especially because it has been shown that gain-of-function mutations in p53 can inactivate ATM.139,140 Conversely, the MDM2 E3 ubiquitin ligase negatively regulates p53 by binding its transcriptional activation domain, inducing its nuclear export, and by polyubiquitinating p53, marking it for proteasomal degradation.141 Abnormal overexpression of MDM2 is found in NSCLC, where it is amplified in a significant number of tumors. MDM2 activity is inhibited by other tumor suppressor genes in the p53 pathway, including ATM and also the p14ARF tumor suppressor gene.142
When p53 is functional, it is activated by phosphorylation in response to cellular stress (e.g., DNA damage). Once activated, p53 strongly induces the expression of other tumor suppressor genes that control cell cycle checkpoints (e.g., p21WAF1/CIP1), apoptosis (BAX), DNA repair (GADD45), and angiogenesis (thrombospondin).143 p53 activation has also been found to alter miRNA expression and maturation.144,145 The high frequency of p53 loss across the entire spectrum of human tumors is a strong testament to its importance in inhibiting tumor development and growth. Restoring p53 activity in tumors is effective at halting their growth and could represent an effective therapy, although development of this strategy is challenging.146 Several gene therapy clinical trials have been reported in which lung cancers are treated by an intratumoral injection introducing a wild-type p53 gene using retroviral or adenoviral vectors.147 Other strategies have been developed to restore p53 activity without attempting to reintroduce the entire gene into tumor cells.148,149 The vaccination of patients with advanced cancer with a custom vaccine corresponding to their tumor’s mutation in p53 or RAS has demonstrated the generation of mutant oncogene–specific immune responses associated with prolonged survival.150 A trial of p53 vaccination in SCLCs produced measurable increases in tumor-specific immune response that was associated with improved outcome and response to therapy in some patients.151,152
CYCLINS AND CELL CYCLE REGULATORY PATHWAYS
p16INK4A is a cyclin-dependent kinase (CDK) inhibitor important for the integrity of the G1 checkpoint. Loss of p16INK4A frees CDKs from inhibition, permitting constitutive phosphorylation of retinoblastoma (RB) protein and inactivation of its growth suppressive function. Approximately 40% of primary NSCLCs lose p16INK4A, located on chromosome 9p21, making it the most common component of the p16INK4A–cyclin D1–CDK4–RB pathway to be inactivated in NSCLC. Other CDK inhibitors are also lost at a lower prevalence in NSCLC, and the RB gene is mutated or lost in a significant minority of cases.153 In contrast, a strikingly different pattern of pathway dysregulation is observed in SCLCs, in which abnormalities in p16 are rarely observed, but RB itself is nearly always abnormal. Although p16INK4A point mutations in NSCLCs were observed in only 14% of tumors, homozygous deletions or aberrant promoter methylation are common mechanisms for p16INK4A inactivation.20 Indeed, aberrant p16INK4A methylation is a frequent, early preneoplastic event in the pathogenesis of SQCC.154 Furthermore, p16INK4A and p14ARF are alternative splice forms of RNA transcripts from the same DNA locus. p14ARF is also a tumor suppressor gene and functions to stabilize p53. Thus, alteration at the p16INK4A locus may not only abrogate p16INK4A function but also disrupt p53 pathway through p14ARF.155
In the absence of inhibitory regulation by the CDK inhibitors, cyclins, and their catalytic partners, the CDKs phosphorylate the retinoblastoma protein, a growth-suppressive nuclear phosphoprotein located on chromosomal region 13q14. When in its active, unphosphorylated form, RB binds and inactivates proteins such as transcription factor E2F-1, preventing G1/S transition. The RB protein is absent or structurally abnormal in more than 90% of SCLCs and 15% to 30% of NSCLCs. Lung-targeted, conditional deletion of RB and p53 in mice leads to the development of SCLC that recapitulate that observed in humans.156 Although the p16 and RB tumor-suppressive components of this pathway are frequently lost in lung cancer, the growth promoting cyclin and CDK components of the pathway are often overexpressed, and cyclin D1, cyclin E1, and CDK4 have each been shown to be amplified in a subset of lung cancers157 and are overexpressed by immunohistochemical evaluation.
LKB1, AMPK, AND MTOR PATHWAY
LKB1 is a serine/threonine kinase that serves as a master regulator
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


