VON HIPPEL-LINDAU DISEASE
Von Hippel-Lindau (VHL) disease is an autosomal-dominant inherited multisystem neoplastic disorder that is characterized by clear cell renal tumors, retinal angiomas, central nervous system hemangioblastomas, tumors of the adrenal gland (pheochromocytoma), endolymphatic sac and pancreatic islet cell, and cysts in the pancreas and kidney. VHL occurs in about 1 in 36,000 and develops during the 2nd to 4th decades of life with nearly 70% penetrance by age 60. Bilateral, multifocal renal tumors with clear cell histology develop in 25% to 45% of VHL patients12 that can have metastatic potential when they reach 3 cm.
Genetics of VHL
Loss of heterozygosity (LOH) on chromosome 3p in clear cell renal tumors suggested the location of a predisposing gene for RCC.13 Positional cloning in VHL kindreds defined the disease locus to chromosome 3p25-26, leading to the cloning of the VHL gene in 1993.5 VHL is a tumor suppressor gene in which both copies of VHL must be inactivated for tumor initiation. Germ-line VHL mutations that predispose individuals to VHL encompass the entire mutation spectrum, including large deletions, protein-truncating mutations, and missense mutations that exchange the amino acid in the VHL protein. Over 700 different VHL mutations have been identified in more than 945 VHL families worldwide. Mutations are located throughout the entire gene with the exception of the first 35 residues in the acidic domain.12 With the development of new methods for detection of deletions, VHL mutation detection rates are approaching 100%.14,15 VHL subclasses based on the predisposition to develop pheochromocytomas and a high or low risk of RCC have been established with clear genotype–phenotype associations emerging.16
Gene Mutated in Renal Cancer Families with Chromosome 3p Translocations
In 1979, Cohen et al.17 described a family with a constitutional t(3;8)(p14;q24) balanced translocation that cosegregated with bilateral multifocal clear cell renal tumors. Loss of the derivative chromosome carrying the 3p segment and different somatic mutations in the remaining copy of VHL were identified in the tumors from this translocation family. Based on these data, Schmidt et al.18 proposed a three-step tumorigenesis model in 3p translocation families: (1) inheritance of the constitutional translocation, (2) loss of the derivative chromosome bearing 3p25, and (3) mutation of the remaining copy of VHL, resulting in inactivation of both copies of VHL and predisposing individuals to clear cell RCC. A number of chromosome 3 translocation families have been described.19,20 Loss of the derivative chromosome concomitant with the somatic mutation of the remaining copy of VHL in these families provides strong evidence for the three-step tumorigenesis model and implicates VHL loss in clear cell RCC that develops in chromosome 3 translocation kindreds.
Gene for Sporadic Clear Cell Kidney Cancer
Somatic mutation of the VHL gene with associated loss of the wild-type allele is found in a high percentage of tumors from patients with clear cell kidney cancer.21 Nickerson et al.22 recently identified mutation or methylation of the VHL gene in 92% of clear cell kidney cancers. The VHL gene mutation is not found in papillary, chromophobe, collecting duct, medullary, or other types of kidney cancer.
Function of the VHL Protein
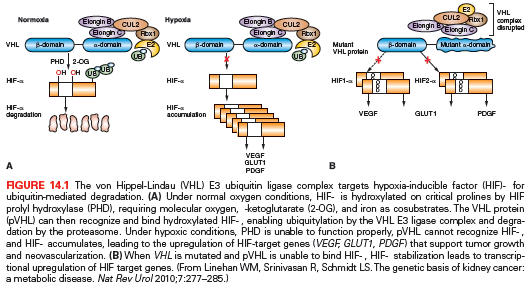
The most well-understood function of the VHL protein pVHL is the substrate recognition site for the hypoxia-inducible factor (HIF)-α family of transcription factors targeting them for ubiquitin-mediated proteasomal degradation (Fig. 14.1).16 pVHL binds through its α domain to elongin C and forms an E3 ubiquitin ligase complex with elongin B, cullin-2, and Rbx-1. Under normal oxygen conditions, HIF-α becomes hydroxylated on critical prolines by a family of HIF prolyl hydroxylases (PHD) that require α-ketoglutarate, molecular oxygen, ascorbic acid, and iron as cofactors. pVHL then binds to hydroxylated HIF-α through its β domain, targeting HIF-α for ubiquitylation by the E3 ligase complex. Under hypoxic conditions when PHDs are unable to function or when pVHL is mutated—thereby altering its binding to HIF-α or elongin C—HIF-α cannot be recognized by pVHL. HIF-α accumulates and transcriptionally upregulates a number of genes important in blood vessel development (EPO, VEGF), cell proliferation (PDGFβ, TGFα), and glucose metabolism (GLUT-1).16 HIF-α–dependent upregulation of target genes involved in neovascularization provides an explanation for the increased vascularity of central nervous system (CNS) hemangioblastomas and clear cell renal tumors in VHL. Germ-line VHL mutations frequently occur in the pVHL binding domains for HIF-α and elongin C.23 HIF-2α (rather than HIF-1α stabilization appears to be critical for renal tumor development.24,25 Additional HIF-independent functions for pVHL have been reported,12,16,26 including an increase of p53 activity through the suppression of MDM2-mediated ubiquitination and nuclear export,27 modulation of nuclear factor κ B (NF-κB) through casein kinase 2 binding and inhibitory phosphorylation of NF-κB agonist CARD9,28 microtubule stabilization,29 maintenance of primary cilium,30 and extracellular matrix formation affecting cell–cell adhesion.31,32
Additional Genes for Clear Cell Kidney Cancer
Studies using next-generation sequencing approaches to identify the genetic basis of clear cell kidney cancer have revealed a considerable number of genetic alterations in chromatin remodeling genes important for the maintenance of chromatin states. Significantly mutated genes identified in sporadic clear cell kidney cancer in addition to VHL include PBRM1, a subunit of the PBAF SWI/SNF chromatin remodeling complex (41%),33 histone methyl transferase SETD2 (4%)34 histone demethylases JARID1C (KDM5C; 3%)35 and UTX (KMD6A; 3%),35 and the novel tumor suppressor gene, BAP1 (15%),36 a histone deubiquitinase. BAP1 is also mutated, albeit rarely, in the germ line of inherited clear cell kidney cancer families37,38 and is associated with poor survival outcome.39
HEREDITARY PAPILLARY RENAL CARCINOMA TYPE 1
HPRC is an autosomal-dominant hereditary cancer syndrome in which affected individuals are at risk for the development of multifocal, bilateral papillary type 1 kidney cancer.40 HPRC develops in the 5th and 6th decades, with age-dependent penetrance estimated at 67% by 60 years of age41; however, early-onset HPRC has been described.42 This rare disorder has been reported in less than 40 kindreds worldwide.40
Genetics of Hereditary Papillary Renal Carcinoma: MET Proto-oncogene
In 1995, Zbar et al.43 described 10 families in which multifocal, bilateral papillary renal tumors were inherited in an autosomal-dominant fashion and suggested that these families might represent a hereditary counterpart to sporadic papillary tumors. Schmidt et al.6 localized the HPRC disease locus to chromosome 7q31.1-34 by genetic linkage analysis. Because the trisomy of chromosome 7 was described as a hallmark feature of papillary renal tumors,44 a gain-of-function oncogene seemed a likely candidate disease gene; in fact, germ-line missense mutations were identified in the tyrosine-kinase domain of the MET protooncogene located at 7q31 in affected HPRC family members.6 Mutations of the MET gene have been detected in 13% of sporadic papillary renal tumors.6,45 Further studies to determine the role of MET and related genes in papillary type 1 kidney cancer are currently under way.
Hereditary Papillary Renal Carcinoma: Functional Consequences of MET Mutations
The MET protooncogene encodes the hepatocyte growth factor/scatter factor (HGF/SF) receptor tyrosine kinase. Binding of ligand HGF to MET triggers autophosphorylation of critical tyrosines in the intracellular tyrosine kinase domain, subsequent phosphorylation of tyrosines in the multifunctional docking site, and recruitment of a variety of transducers of downstream signaling cascades that regulate cellular programs leading to cell growth, branching morphogenesis, differentiation, and “invasive growth.”46 Although MET overexpression has been demonstrated in a number of epithelial cancers,47 HPRC was the first cancer syndrome for which germ-line MET mutations were identified. The missense MET mutations in HPRC are constitutively activating without ligand stimulation, display oncogenic potential in vitro,48,49 and are predicted by molecular modeling to stabilize active MET kinase.50 Nonrandom duplication of the chromosome 7 bearing the mutant MET allele was demonstrated in papillary renal tumors from HPRC patients51 and may represent the second step in HPRC tumor pathogenesis. The presence of two copies of mutant MET may give kidney cells a proliferative growth advantage and lead to tumor progression.
XP11.2 TRANSLOCATION RENAL CELL CANCER
Xp11.2 translocation renal cell carcinomas, typically presenting with papillary architecture and clear or eosinophilic cytoplasm, are rare tumors in adults (1.6%), but are the cause of 20% to 45% of renal cancers in children and young adults. Translocations involving Xp11.2 and 1q21.2 associated with sporadic papillary renal carcinoma, and first described in a 2-year-old child,52 generate a fusion between a novel gene, PRCC, and the basic helix-loop-helix-leucine zipper transcription factor gene, TFE3, a member of the microphthalmia (MiT) family of transcription factors.53 The encoded fusion protein, PRCC-TFE3, acts as a stronger transcriptional activator than native TFE3, and a loss of the majority of native TFE3 transcripts is observed in these tumors. This deregulation of normal TFE3 transcriptional control caused by the chromosomal translocation may be important to the development of sporadic papillary renal cell carcinoma.54,55 Xp11.2 translocation renal cell carcinomas involving at least five different TFE3 gene fusions and resulting in deregulation of TFE3 transcription activity have been described, including NonO-TFE3, PSF-TFE3, CLTC-TFE3, and ASPL-TFE3.56 Tsuda et al.57 have shown that these TFE3 fusion proteins are strong transcriptional activators of the MET gene, resulting in inappropriate MET-directed cell proliferation and invasive growth. Given the physiologic consequences of TFE3 fusion protein expression, therapeutic targeting of MET may be an effective treatment for Xp11.2 translocation renal tumors.
The fusion of another member of the MiT family, TFEB, with the Alpha gene has been described in renal tumors harboring t(6;11)(p21;q13) chromosomal translocation.58 The first case of renal cancer involving a third MiT family member, MITF, was recently reported, in which an activating MITF mutation was identified in the germ-line of family members affected with renal tumors.59
BIRT-HOGG-DUBÉ SYNDROME
BHD syndrome is a rare autosomal-dominant inherited cancer syndrome characterized by benign tumors of the hair follicle (fibrofolliculoma), pulmonary cysts and spontaneous pneumothorax, and a sevenfold increased risk for renal cancers.60–63 Fibrofolliculomas and lung cysts are the most common manifestations (>85%) of BHD patients.64–66 Renal tumors with variable histologies develop in about 30% of BHD-affected individuals (median age, 48 to 50 years), most frequently chromophobe renal carcinoma and hybrid oncocytic tumors.64,67 Metastases may develop from BHD renal tumors, but they are uncommon.
Genetics of Birt-Hogg-Dubé Syndrome: Folliculin Gene
A genetic linkage analysis performed in BHD kindreds led to the localization of the disease locus on the short arm of chromosome 1768,69 and the identification of the BHD gene, FLCN.8 Almost all BHD-associated FLCN mutations are predicted to truncate the BHD protein, folliculin, including insertion or deletion, nonsense, and splice-site mutations,8,64,65,70 but recently, several missense mutations located in conserved amino acid residues and partial gene deletions have been described.65,71–73 The mutation detection rate in several large BHD cohorts approached 90%, and germ-line mutations were distributed throughout the entire length of the FLCN gene with no clear genotype–phenotype correlations.64,65,74 Vocke et al.75 identified second “hit” somatic mutations or LOH in 70% of renal tumors from BHD patients, supporting a role for FLCN as a tumor suppressor gene that predisposes individuals to renal tumors when both copies are inactivated. FLCN mutations have been found infrequently in chromophobe renal cell carcinomas (11%),76 and only rarely in other histologic variants of RCC.77–79
Function of the Birt-Hogg-Dubé Protein: Folliculin
The FLCN gene encodes a novel protein, folliculin (FLCN), with no characteristic functional domains. Baba et al.80 identified a novel FLCN-interacting protein, FNIP1, and showed that FNIP1 interacts with the γ subunit of 5′ adenosine monophosphate (AMP)–activated protein kinase (AMPK), an energy sensor in cells that negatively regulates mammalian target of rapamycin (mTOR), the master switch for protein translation and cell proliferation, through TSC1/2.81,82 A second folliculin interacting protein, FNIP2, was subsequently identified that displayed similar biochemical properties to FNIP1.83,84 FLCN, through FNIP1/2, may play a role in the regulation of the AMPK-TSC1/2–mTOR signaling pathway (Fig. 14.2). Published data from in vivo models and BHD renal tumors, supporting mTOR activation85,86 as well as mTOR inhibition87–89 as a consequence of FLCN inactivation, has led to the hypothesis that the mechanism by which FLCN interacts with and modulates mTOR is context dependent.89
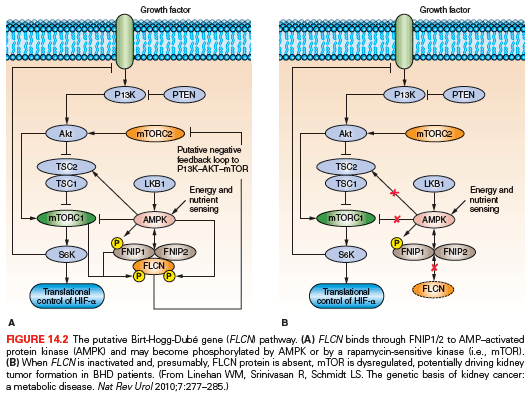
Additional functional roles for FLCN in transforming growth factor β (TGF-β) signaling,90,91 modulation of HIF-α and its target genes,92 ciliogenesis,93 peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) regulation and mitochondrial biogenesis,94,95 cell–cell adhesion,96,97 exit from embryonic stem cell pluripotency,98 and regulation of lysosome function through cytoplasmic sequestration of the transcription factors TFE399 and TFEB100 have been reported. The resolution of C-terminal FLCN crystal structure has demonstrated structural homology to the DENN domain, which has guanine exchange factor (GEF) activity toward RabGTPases.101 Two recent reports have shown FLCN/FNIP1/FNIP2 interaction with RagGTPases and proposed a role of this complex in amino acid sensing for mTOR activation.100,102 In addition to a role for FNIP1 in facilitating FLCN interaction with AMPK, Fnip1 knockout mice displayed a defect in pro- to preB cell differentiation demonstrating a unique requirement for the FLCN-FNIP1 complex in B-cell development.103
HEREDITARY LEIOMYOMATOSIS AND RENAL CELL CARCINOMA
HLRCC is an autosomal-dominant inherited disorder that predisposes individuals to the development of skin and uterine leiomyomas and an aggressive form of type 2 papillary renal carcinoma. Fewer than 150 HLRCC families have been reported worldwide.104,105 Renal tumors, which are often unilateral and solitary,104,106 may develop with early age of onset in 15% to 25% of affected individuals104,105
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


