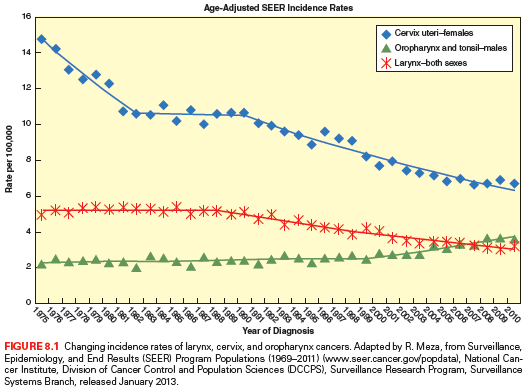
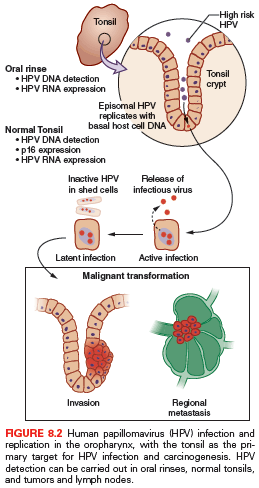
HPV is the most common sexually transmitted disease with an overall genital HPV prevalence of 42.5% in females 14 to 59 years of age as determined in the National Health and Nutrition Examination Survey (NHANES) (2003–2006).23 In the oral cavity and oropharynx, the lymphoid-rich tissue of the tonsils is the likely reservoir of HPV. To replicate, HPV must infect the basal epithelial cell. The thin epithelium of the tonsillar crypts serves as an ideal location for the virus to infect (Fig. 8.2). HPV infection may be facilitated by concurrent inflammation within the tonsil due to other microorganisms. D’Souza et al.24 conducted a hospital-based case control study of 100 patients with newly diagnosed oropharynx cancer to 200 noncancer patients to evaluate associations between HPV infection and oropharyngeal cancer. A high lifetime number of vaginal sex partners (≥26) and oral sex partners (≥6) was significantly associated with oropharyngeal cancer as was seropositivity for antibodies to the viral L1 protein or oral infection with any hrHPV type, most frequently HPV16. However, other high-risk HPV types are also found in roughly 10% of oropharynx tumors and in other head and neck cancer locations, notably the nasopharynx and the oral cavity; the proportion of other high-risk HPV types is much higher.25,26 Oral and vaginal HPV infections in a series of HIV-positive and HIV-negative women revealed a significant but lower incidence of oral HPV (15%) compared to vaginal HPV (51%).27 Oral HPV infection incidence in the general U.S. population was evaluated in the NHANES study.28 The prevalence of oral HPV infection shows a biphasic distribution with one peak in the 30- to 34-year-old age group and a second peak in the 60- to 64-year-old age group. Overall, oral HPV infection was (6.9%), with high-risk HPV types in 3.7%. Men had a higher prevalence than women (10.1% versus 3.6%), and blacks had a higher prevalence of oral HPV (10.5%) than whites (6.5%), although this difference did not reach statistical significance. It is not yet certain how oral HPV infection relates to the risk of developing HPV-induced head and neck cancer.
Fortunately, HPV-positive oropharyngeal cancers show improved response to therapy when compared to HPV-negative cancers of the same site.29–32 Patients presenting with HPV-positive oropharyngeal cancers also tend to be younger, healthier, and have a much lower frequency of smoking and alcohol abuse.33 However, some studies have shown that a history of smoking in patients with HPV-positive oropharynx cancer may increase the likelihood of recurrence or metastasis after treatment.21,34 This has prompted many to consider reducing the intensity of treatment to decrease treatment-related morbidity.35,36 In contrast to the excellent prognosis of HPV-positive oropharynx cancers, in HPV-positive oral cancers,37 and HPV-positive nasopharyngeal cancers,38 the outcome may be worse than HPV-negative cancers arising in these sites.
At the University of Michigan, 80% to 90% of oropharyngeal tumors (see Table 8.1) that present are HPV positive,21,25 and >80% of patients with HPV-positive oropharynx cancer are responders to concurrent carboplatin and taxol with intensity-modulated radiation treatment (chemoRT).39 Critical questions for the future are to determine which patients can benefit from less intensive treatment, which patients need the intensive concurrent chemoRT treatment, and which patients need a different treatment because they fail to be cured by chemoRT. Spector et al.40,41 showed that nodal status and, in particular, matted nodes (defined as three nodes abutting one another with loss of an intervening fat plane replaced with evidence of extracapsular spread) defines a group of patients with a greatly increased risk of distant metastasis and poor overall and disease-free survival. The molecular correlates of these highly malignant HPV-positive tumors are yet to be defined fully, but in work submitted for publication, Walline et al. have shown that integration of hrHPV into cancer-related genes is common in tumors of patients who suffer from recurrent disease and poor survival, whereas integration of the viral genome into intragenic sites was more commonly associated with good response to therapy and prolonged progression-free survival. Immune responses to HPV antigens have also been suggested to be a cause of the good response to therapy among HPV-positive oropharyngeal cancers. However, failure of a potent immune response in some cases may occur because of frequent alterations affecting the tumor necrosis factor (TNF) receptor TRAF3, which participates in the activation of the immune response and nuclear factor kappa B (NF-κB) signaling (TCGA Head and Neck Cancer Consortium).42 These changes may provide a mechanism for HPV-positive tumors to evade antiviral immune responses.42 As a group, HPV-positive head and neck cancers have fewer gross genetic changes than HPV-negative tumors and share specific similarities, such as wild-type TP53 and intact p16ink4a (CDKN2A), both of which are rare in HPV-negative HNSCC. Nearly all HPV-positive HNSCC express the HPV E6 and E7 viral oncogenes and exhibit low or absent expression of the HPV E2 gene that serves as a negative regulator of E6 and E7 expression (Walline et al. submitted). Viral integration43 appears to be a requirement for carcinogenesis. Integration typically disrupts E2, which results in the unregulated expression of the E6 and E7 oncoproteins and includes the expression of the alternate HPV E6 transcripts E6*I and E6*II.44 It appears that viral integration also causes a significant disruption of the host genome at sites of viral integration.45 When significant disruption occurs leading to other genomic damage, this could also be a factor in the subset of nonresponsive HPV-positive tumors.
MOLECULAR MECHANISMS IN HEAD AND NECK SQUAMOUS CELL CARCINOMA
Early molecular studies of head and neck cancers revealed that amplification of the epidermal growth factor receptor (EGFR) gene with associated EGFR overexpression are common among head and neck cancers and are associated with poor prognosis.46,47 It is puzzling that only about 10% of HNSCC cases respond to treatment that targets EGFR.48 Alternate driver pathways are suspected as an explanation for this observation. Early molecular studies of HNSCC also revealed that mutations, loss of heterozygosity, and methylation of the CDNK2A locus is very common and occurs early in HNSCC development.49–51 Mutation and overexpression of p53 was also identified in early studies of HNSCC.52 CyclinD1 amplification and overexpression has also been identified as an important biomarker that is associated with outcomes and responses to treatment.53 In the landmark VA larynx trial, a biomarker analysis revealed that overexpressed p53 predicted the response to induction chemotherapy and radiation (RT).54 Because p53 overexpression is nearly always associated with mutant p53, this suggested that tumors with wild-type p53 were less responsive to chemotherapy and RT, even though normal cells containing wild-type p53 are very sensitive to chemotherapy or RT. Thus, it was unclear why tumors with wild-type p53 were more resistant to chemotherapy and RT. Subsequent studies revealed that the combination of wild-type p53 and overexpression of Bcl-xL resulted in cisplatin resistance.55 This suggested that Bcl-xL blocks p53-induced apoptosis, and that wild-type p53 mediates cell cycle arrest and repair mechanisms, allowing tumor cells to escape from treatment-induced damage.
In spite of our awareness of EGFR overexpression and its potential value as a target for therapy, it is still impossible to predict which head and neck cancers will respond and which will not. The same is true for many other molecular markers when only one molecular target is addressed. It is now becoming clear that it will be necessary to identify the driver pathways and target more than one cancer driver. It was postulated many years ago that a certain number of “hits” is necessary to drive a tumor. Knudsen correctly explained the tumor suppressor nature of the retinoblastoma gene by comparing the kinetics of tumor development in children with an inherited predisposition and in children who developed sporadic retinoblastoma. When time to tumor development in affected children was plotted against months from birth, there was a straight line for children who inherited a mutant allele from an affected parent. However for children with sporadic retinoblastoma the line had a plateau for several months and then fell linearly. From this he reasoned that two hits were necessary for tumor development. For children who inherited one normal and one mutant gene he observed first-order kinetics, and for children who did not inherit a mutant gene he observed second-order kinetics. Thus only one additional event was necessary for tumor development in the inherited disease, but two events were necessary in the sporadic cases, i.e. acquisition of a mutation in a cell and then a second event in the same cell, resulting in loss of the healthy gene. For tumors that arise in adult epithelial tissues, the situation is more complex. It was estimated many years ago using a similar mathematical approach in cancers that develop with aging that it is likely that four to five events are necessary for a tumor to develop.
To attempt to identify cancer drivers, we characterized chromosomal abnormalities in two cell lines derived from a patient with recurrent laryngeal cancer. One portion of the tumor was within the endolarynx. From this, the UM-SCC-17A cell line was established, and from a second portion of the tumor that had invaded through the thyroid cartilage into the soft tissues of the neck, UM-SCC-17B was established. There were a total of 22 chromosome rearrangements in the two cell lines, of which 8 were unique to the UM-SCC-17A cell line from the endolarynx and 9 were unique to the UM-SCC-17B cell line from the invasive lobe of the tumor. Of interest, however, were the five rearrangements that were common to both cell lines, which could represent the loci of genetic events associated with tumor initiation, whereas the others represent either random events or events associated with progression.56 It will be of interest to use modern sequencing methods to identify the genetic aberrations linked to these five early chromosome rearrangements to test this hypothesis. Hahn and Weinberg tested what steps were necessary to transform normal human bronchial epithelial cells into tumor cells. They targeted a small but consistent set of specific pathways based on the observed abnormalities of many epithelial cancers. The necessary pathways include: ectopic expression of the catalytic unit of human telomerase, human telomerase reverse transcriptase (hTERT), to stabilize telomeres over many cell divisions; abrogation of the Rb pathway with SV40 large T oncoprotein to induce continuous cell cycle entry; inhibition of TP53 with large T oncoprotein to facilitate cell cycle progression and to inhibit TP53-mediated induction of apoptosis; and introduction of the activated (oncogenic) allele of HRAS, to provide continuous signaling through the RAS, RAF, mitogen-activated protein kinase (MEK), extracellular signal-related kinase (ERK) kinase cascade to trigger the expression of transcription factors associated with proliferation.57,58
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


