THE RAS-RAF-MAP KINASE PATHWAY
The RAS-RAF–mitogen-activated protein kinase (MAPK) signaling pathway is a cascade of molecules that is activated by multiple cellular signals and pathways (Fig. 21.2). Signaling through RAS and RAF leads to activation of the ERK1/2 kinases, which regulate a variety of proteins through serine-threonine phosphorylation events and ultimately the transcription of many genes governing cell proliferation, survival, and other critical cellular processes. Examples of transcription factors operant in melanocytes that are regulated by ERK signaling include the microphthalmia-associated transcription factor (MITF, described in detail in the following), various ETS transcription factors, the FOS and JUN immediate early genes, among others.
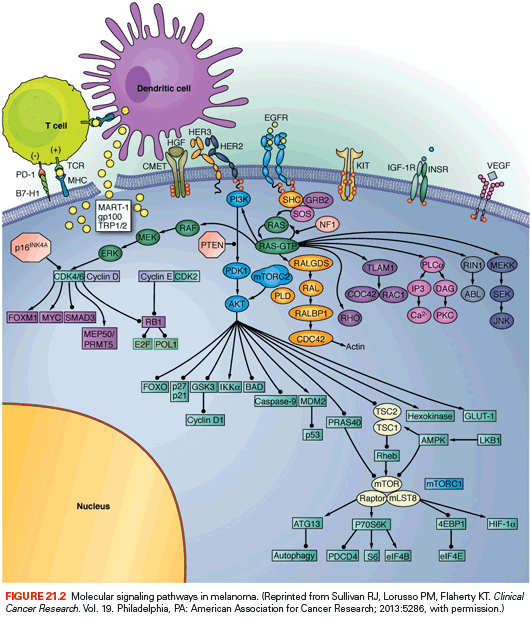
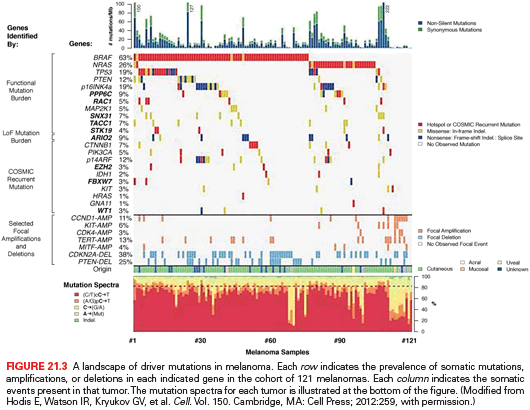
Extensive genetic and mechanistic studies have unearthed a prevalence of activating MAPK pathway mutations across many tumor types. In particular, activation of this pathway appears to be one of the most frequent and important molecular events in cutaneous melanoma (Fig. 21.3). Toward this end, several MAPK signaling proteins (e.g., RAS and RAF isoforms) are encoded by “classic” oncogenes, and key transcriptional effectors downstream of MAPK also undergo oncogenic dysregulation in melanoma and other cancers. Key MAPK effectors have also been shown to regulate differentiation and senescence in non-transformed melanocytes.
RAS Family GTPases
RAS proteins (H-, K-, and NRAS) are small GTPases that comprise an initial signaling node of the RAS-RAF-MAPK cascade. The discovery of activating mutations in HRAS and KRAS led to investigations that identified mutations in this gene family in multiple cancer types, and thus the significance of this pathway in the pathogenesis of cancer. Activating mutations in NRAS are detected in 20% to 25% of cutaneous melanomas (see Fig. 21.3).10,11,12 NRAS mutations are also detected in nevi, particularly congenital nevi.13 HRAS mutations are uncommon in cutaneous melanomas, but they are detected in Spitz nevi, which are rare, benign lesions most often diagnosed in children and young adults.14 Despite their high incidence in other cancer types, KRAS mutations are extremely rare in melanocytic lesions.
In mouse models, overexpression of activated HRAS or NRAS on an Ink4a/Arf-null background results in spontaneous melanoma formation.15,16 However, while HRAS-induced melanomas rarely, if ever, metastasize, NRAS tumors frequently metastasize to draining lymph nodes and distal organs, in line with the apparent selection for NRAS over HRAS mutations in human melanomas. Knockdown of NRAS in human melanoma cell lines inhibits their viability, indicating dependency on this oncogene for tumorigenicity.17 Furthermore, shutting off transgene expression in an inducible NRAS model caused regression of melanomas that arose following transgene induction, thereby confirming the RAS oncogene dependency in these tumors.18
RAF Kinases
The RAF proteins (ARAF, BRAF, and CRAF) are serine-threonine kinases that comprise critical signaling effectors through the RAS-RAF-MAPK pathway (see Fig. 21.2). While each of these proteins likely plays a role in physiologic signaling, BRAF has a central role in the pathogenesis of melanoma. Somatic hotspot mutations in BRAF are detected in 40% to 45% of cutaneous melanoma, making them the most common oncogenic aberration detected in this disease to date.10 Approximately 95% of these mutations result in substitutions of the valine at the 600 position in the protein. The most common mutation (~70% of BRAF mutations) is a T→A transversion, resulting in a valine to glutamate amino acid substitution (V600E). Although the T→A transversion is not classically associated with UV-induced damage, BRAF V600E mutations appear are more common in melanomas arising at sites with intermittent exposure to UV.19 Other substitutions, particularly the V600K mutation that represents ~20% of BRAF mutations in melanoma, are more common in melanomas with evidence of chronic sun damage (CSD), although the overall rate of BRAF mutations in those melanomas is lower compared to tumors without CSD.19–22 BRAF V600 mutations are an early event in melanomas, as they are also present in the majority (~80%) of benign and dysplastic nevi.23 In addition to mutations affecting V600, somatic events affecting >20 other sites in BRAF have been detected in patients, but overall they are quite rare (total ~5% prevalence).24
BRAF is an immediate downstream target of RAS (see Fig. 21.3) in the MAPK pathway. The BRAF(V600E) mutation and other substitutions at the V600 site confer more than 200-fold induction of kinase activity in vitro.25 Mutations affecting other sites in BRAF can have high, intermediate, or low catalytic activity. However, all of these mutations cause increased activation of MEK and ERK signaling. This likely occurs in low-activity mutants due to conformational changes that promote heterodimer formation with other RAF isoforms, such as CRAF, in a multiprotein complex with RAS proteins.24 Notably, while BRAF(V600) mutations and NRAS mutations are mutually exclusive in newly diagnosed melanomas, frequent co-occurrence of NRAS mutations with nonactivating BRAF mutations has been observed.26,27
Extensive data suggest that wild-type BRAF operates on a senescence pathway in benign human nevi. Transgenic expression of BRAF(V600E) targeted to melanocytes in zebrafish produced benign nevus-like lesions, whereas invasive melanomas were produced (after extended latency) when crossed into p53-deficient zebrafish.28 Inducible expression of BRAF(V600E) alone in murine melanocytes resulted in excessive skin pigmentation and the appearance of nevi containing hallmarks of senescence.29 Human congenital nevi with activating BRAF mutations were shown to express senescence-associated acidic β-galactosidase, the classical senescence-associated marker.30 This implied that activated BRAF alone is insufficient to induce tumor progression beyond the nevus stage in patients. Interestingly, immunohistochemical staining of nevoid tissues found heterogeneous patterns of INK4A that only partially overlapped with senescence-associated acidic β-galactosidase, suggesting the presence of INK4A-independent pathway(s) operative in oncogene-induced senescence. Expression of BRAF(V600E) in murine melanocytes, in the setting of inactivation of INK4A, caused melanocyte hyperplasia, but no invasive lesions.31 However, concurrent loss of phosphatase and tensin homolog (PTEN), a negative regulator of the phosphatidylinositol kinase (PI3K) signaling pathway, resulted in 100% penetrance of invasive melanomas that formed spontaneous metastases.
The significance of BRAF mutations is now also supported by the functional and clinical effects of inhibiting this target. Early experiments demonstrated that RNAi knockdown of BRAF in human melanoma cells with BRAF(V600E) mutations inhibits ERK activation, induces cell cycle arrest and/or apoptosis, and blunts cell growth.32,33 These initial results led to the development and testing of potent and selective inhibitors of the BRAF(V600E) protein. Two of these agents (vemurafenib, dabrafenib) have demonstrated significant improvements in patient outcomes in phase 3 clinical trials, leading to their regulatory approval, while other agents have shown promising results in early testing.34,35
Interestingly, use of these selective inhibitors of BRAF(V600E) causes paradoxical activation of the MAPK pathway in melanomas with a wild-type BRAF, particularly in those tumors that have an activating NRAS mutation.36,37 This pathway activation causes increased tumor growth in cell lines and xenograft models, and supports the restricted use of these inhibitors to patients with melanoma with activating BRAF mutations. This paradoxical signaling effect also appears to be the underlying mechanism of the proliferative cutaneous lesions (keratoacanthomas and squamous cell carcinomas) that are among the most common toxicities of the selective BRAF inhibitors.38,39
While only rarely mutated in melanoma, preclinical studies support the premise that CRAF can also have functional significance in this malignancy. Cells with activating NRAS mutations appear to utilize CRAF predominantly to transmit signals to MEK and ERK.26 While CRAF appears to be largely dispensable in melanomas with BRAF(V600) mutations, it is likely critical for pathway activation through heterodimer formation with nonactivating BRAF mutations, and it may be a therapeutic target in such tumors.40 Increased expression and/or signaling by CRAF has also been implicated as a mechanism of resistance to mutant-selective BRAF inhibitors in melanomas with BRAF(V600E) mutations.41–43
MEK1/2
Because the MEK1/2 serine-threonine kinases transmit the critical MAPK signal downstream of RAS and RAF, considerable interest has also emerged regarding these kinases in melanoma biology and therapeutics. MEK1/2 mutations are rare in melanoma, and in cancer in general, in contrast to RAS and BRAF mutations.11,12,44 Nonetheless, pharmacologic MEK inhibition presents another possible therapeutic strategy for BRAF– or NRAS-mutant melanomas. Robust preclinical evidence favoring this notion derived from a genetic and pharmacologic analysis showing that various MEK inhibitory compounds demonstrated markedly enhanced potency against BRAF(V600E) cancer cells compared to cell lines lacking oncogenic MAPK pathway mutations.45 Treatment with the MEK1/2 inhibitor trametinib as a single agent in patients with metastatic melanoma with BRAF(V600E) or BRAF(V600K) mutations resulted in significant improvements in clinical response rates, progression-free survival, and overall survival compared to chemotherapy, leading to its regulatory approval in 2013.46 In addition, combined treatment with selective RAF inhibitors (i.e., dabrafenib) and MEK inhibitors (i.e., trametinib) significantly improves both the rate and duration of clinical responses.47 This pivotal observation led to the approval of combined RAF/MEK inhibition for use in BRAF(V600E)-mutant melanoma. Interestingly, this combination also has less cutaneous toxicity than is observed with single-agent BRAF inhibitors, likely due to blockade of the paradoxical activation of RAS-RAF-MAPK pathway signaling in keratinocytes with RAS mutations.39
While mutant-selective BRAF inhibitors are contraindicated for patients with activating NRAS mutations, preclinical studies have demonstrated that at least some NRAS-mutant melanoma cells were also sensitive to MEK inhibitors.45 Recently the MEK1/2 inhibitor MEK162 has demonstrated promising results in patients with metastatic melanoma with NRAS mutations, leading to randomized clinical studies versus chemotherapy.48 MEK inhibitors have also demonstrated efficacy in NRAS-mutant melanomas as a part of combinatorial strategies (i.e., with PI3K inhibitors, CDK4 inhibitors, etc.). The safety and efficacy of several of these combinations in patients are currently being investigated in clinical trials.49
NF1
A small percentage of melanomas lack BRAF or NRAS mutations; however, MAPK signaling is often still operant in this setting. Recent genome characterization efforts have revealed that the tumor suppressor gene NF1 undergoes inactivating mutations in a substantial proportion of “BRAF/NRAS wild-type” melanomas.11,12 NF1 encodes neurofibromin, a so-called RAS-GAP whose normal physiologic role involves negative regulation of RAS signaling effected through cleavage of the RAS-GTP. Consequently, loss of NF1 leads to dysregualted RAS signaling. NF1 loss is sufficient to drive melanoma genesis together with other known cancer genes in genetically engineered mouse models of melanoma.50 Moreover, NF1 loss has been observed in BRAF-mutant clinical specimens that exhibit resistance to RAF inhibition.50,51
THE MEPK PATHWAY AND THERAPEUTIC RESISTANCE
Unfortunately, the impressive clinical effects of RAF and MEK inhibition in BRAF(V600)-mutant melanoma are transient: the vast majority of patients will experience disease progression within one year of treatment (although a subset of patients may enjoy prolonged clinical benefit). Multiple mechanisms of resistance to RAF/MEK inhibition have been described, most of which restore MEK/ERK signaling as the key downstream mechanism. Resistance mechanisms such as NRAS mutations52 or NF1 inactivation51 accomplish this by signaling through C-RAF, which is normally inactive in BRAF-mutant melanoma but can be engaged through homo- or heterodimerization in the setting of upstream RAS signaling. An alternative splice isoform of BRAF also acts through RAF-mediated dimerization.43 Certain receptor tyrosine kinase–driven resistance mechanisms may also work in part by augmenting A- and/or C-RAF activity in a RAS-dependent manner.42,53 Other resistance effectors may bypass RAF proteins altogether yet still converge on MEK/ERK activation—the kinase COT (encoded by the MAP3K8 gene) comprises an example of this mechanism.54 Alternatively, MEK/ERK signaling may be restored through activating somatic mutations in MAP2K1 or MAP2K2, which encode MEK1 and MEK2 kinases, respectively.55,56 Interestingly, NRAS and MAPK2 mutations have been observed in the setting of resistance to both single-agent RAF inhibition and combined RAF/MEK inhibition,57 underscoring the importance of sustained ERK signaling as a resistance mechanism to this therapeutic modality.
Although MEK/ERK-independent resistance mechanisms are less well understood, recent data suggest that such effectors may also prove important. For example, a large-scale functional screen for open reading frames whose overexpression produces resistance to MAPK inhibitors identified several dozen genes that can confer resistance to RAF, MEK, and ERK inhibition.58 Several of these genes encode proteins that may bypass MEK/ERK signaling altogether, whereas others encode transcription factors known to operate downstream of ERK (such as MITF) or that can otherwise substitute for the ERK-driven transcriptional output. As combinatorial MAPK-directed therapy gains traction in BRAF(V600)-mutant melanoma, such ERK-independent resistance mechanisms are likely to become increasingly manifest.
Cell Cycle Regulators
The RB signaling pathway regulates the entry in and progression through the cell cycle. A significant role for this pathway in melanoma was initially implicated by the finding that germline mutations in this pathway (CDKN2A, CDK4) are the most frequently detected events in familial melanomas (more than three affected family members). Subsequent studies have demonstrated that somatic aberrations in this pathway are ubiquitous in cutaneous melanoma. Functional studies support that dysregulation of cell cycle entry and progression is critical to the pathogenesis of this disease, and potentially contributes to resistance to RAS-RAF-MAPK pathway inhibitors.
The CDKN2A Locus
Germline deletions and activating mutations in the CDKN2A locus on chromosome 9p21 are the most common event (~40%) prevalent in familial melanoma. Somatic mutations and deletions, or epigenetic silencing, are detected in as many as 70% of cutaneous melanomas.59,60 Thus, disruption of CDKN2A function likely plays a central role in melanoma pathogenesis. The CDKN2A locus contains an unusual gene organization, which allows for two separate transcripts and corresponding tumor suppressor gene products to be produced: p16INK4A and p19ARF (see Fig. 21.2). Loss of p16INK4A results in the suppression of retinoblastoma (RB) tumor suppressor activity via increased activation of the CDK4/6-cyclin D1 complex; loss of ARF (p14ARF in human and p19ARF in mouse) downmodulates p53 activity through increased activation of MDM2. Thus, deletion of the entire locus accomplishes the inactivation of two critical tumor suppressor pathways: RB and p53. Homozygous deletion of exons 2 and 3 of the mouse Cdkn2a homolog predisposed to a high incidence of melanomas when combined with an activated HRAS transgene in melanocytes.15 Thus, CDKN2A lesions may “prime” melanocytic tissue for neoplasia.
INK4A
The specific significance of INK4A function is supported by the clinical identification of intragenic mutations of INK4A that do not affect the ARF coding region that sensitize germline carriers to the development of melanomas.61 In addition, in familial melanomas that lack CDKN2A aberrations, the most commonly identified genetic event is a point mutation in CDK4 that disrupts the interaction of that protein with INK4A.62 In a mouse model engineered to be deficient only for Ink4a (with intact ARF), melanoma formation was observed in cooperation with an oncogenic initiating event (activated HRAS), albeit with a longer latency than in mice with deletions affecting the entire locus.63 Notably, the tumors in these mice were also found to harbor either deletion of ARF or mutation of p53. Therefore, while INK4A is a bona fide tumor suppressor, additional genetic dysregulation of the p53 pathway seems obligatory for melanoma genesis, at least in the mouse.
CDK4
CDK4 is a direct target of inhibition by p16INK4A (see Fig. 21.2) and is a primary regulator of RB activation. As noted previously, germline mutations of CDK4 that render the protein insensitive to inhibition by INK4A (e.g., Arg24Cys) have been identified in a melanoma-prone kindreds.61 These tumors retain wild-type INK4A function, suggesting that INK4A is epistatic to CDK4 and that RB pathway deregulation is central to melanoma genesis. Somatic focal amplifications of CDK4 are also observed (albeit rarely) in sporadic melanomas.64 Carcinogen treatment induced melanomas in the animals without somatic Ink4a inactivation, similar to the mutual exclusivity observed in familial melanoma.65 CDK4 interacts with cyclin D proteins (see the following) to drive progression through the G1/S cell cycle checkpoint. Recently, small molecule inhibitors of CDK4 and other CDKs have entered clinical trials in several tumor types, including melanomas. In particular, both tumor genetic data and recent results from genetically engineered mouse models provide a rational basis for combining CDK and MAPK pathway inhibition in NRAS-mutant melanoma.18
CCND1
CCND1 encodes the CyclinD1 kinase, which forms a complex with CDK4 or CDK6 to inactivate RB1 (see Fig. 21.2). Amplification of the CCND1 locus has been identified in relatively rare event in cutaneous melanomas (5% to 10%).11,66 However, this molecular event is enriched in cutaneous melanomas that do not have mutations in BRAF or NRAS.11,19 Although it is rare in melanomas with BRAF(V600) mutations, amplification of CyclinD1 has been implicated as predictor of resistance to BRAF inhibitors in preclinical models, and increased copy number of CCND1 correlated with shorter progression-free survival in one study of patients with metastatic melanoma treated with the BRAF inhibitor dabrafenib.67,68
RB1
Germline mutations in RB1 confer predisposition to melanoma in patients who have survived bilateral RB.69 These melanomas exhibit loss of heterozygosity of the remaining wild-type RB1 allele. In such patients, estimates of increased lifetime risk of melanoma range from 4- to 80-fold. The RB1 gene locus has been found deleted in some primary cutaneous melanomas,13 and RB1 may also be subject to genomic rearrangement in rare instances.14
The p53 Pathway
The p53 pathway is critical for maintenance of the normal genome by regulating a multiplicity of mechanisms, including cell cycle checkpoints, DNA damage repair activation, and the appropriate induction of apoptosis. Mutations in the TP53 gene occur in >50% of all tumors. While initial studies suggested that the TP53 locus is rarely mutated in human melanomas, whole exome studies have identified mutations in ~20% of tumors, generally in tumors without mutations or deletions affecting CDKN2A and P14ARF.11 Amplification of MDM2, which inhibits P53 function, has also been detected in melanomas with intact CDKN2A.64 Functionally, loss of p53 cooperates with activated BRAF in zebrafish, and with activated HRAS in mice, to induce melanomas.28,70 Thus, while TP53 is rarely deleted in human melanomas, inactivation of its pathway appears critical for melanomagenesis.
The Phosphatidylinositol 3-Kinase Pathway
The PI3K-AKT pathway is affected by activating oncogenic events more frequently than any other pathway in cancer.71
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


