Global Events in Colorectal Cancer
CRCs acquire genetic instability in stereotypic ways that favor the accumulation of hundreds to thousands of somatic aberrations (see Fig. 13.1). About 80% of tumors display widespread chromosomal gains, losses, and translocations—phenomena that lead to gene amplifications, rearrangements, and deletions.10 These tumors carry, on average, below 100 somatic nonsynonymous point mutations. Chromosomal segregation defects may account for chromosomal instability (CIN), as the segregation factor Bub1 illustrates in mice,11 but few specific gene defects are implicated confidently. Beyond weak associations with structural changes on chromosomes 8 and 18,12 specific cytogenetic features barely influence disease patterns or patient outcomes.
About 15% of CRCs appear globally euploid but carry thousands of point mutations and small deletions or insertions near nucleotide repeat tracts—the defect designated as MSI-hi.13 Features and the molecular determinants of disease progression in MSI+ adenomas differ from those associated with CIN; for example, BRAF V600E mutations are more common in MSI+ precursor adenomas than in other types.14 Because hypermutability, whether associated with CIN or MSI, results in many changes that are inconsequential or even detrimental to tumors, the mere presence of a mutation does not signify a pathogenic role. Therefore, two features are used to distinguish driver from passenger mutations: appearance in a high fraction of tumor specimens, and ideally, the experimental demonstration of its contribution to a malignant property.
Epigenetic mechanisms may be as significant as mutations in cancer but are less well understood. Various covalent histone modifications and methylation of cytosine residues in DNA represent prominent modes of gene regulation,15 the latter being far better characterized in CRCs than the former. 5′-CpG-3′ dinucleotide pairs are particular targets for methylation in localized areas of high CpG content in promoters, where methylation silences adjacent genes. CRCs show 8% to 15% lower total DNA methylation than normal tissue,16 even in colorectal adenomas.17 Reduced pericentromeric methylation might decrease the fidelity of chromosomal segregation and altered methylation or loss of imprinting at the IGF2 locus increase CRC risk,18 suggesting broad effects of global hypomethylation on cell growth. However, its precise significance is unclear because some animals show increased tumor susceptibility with global hypomethylation,19 whereas ApcMin mice lacking or overexpressing the de novo DNA methyltransferase DNMT3B show reduced or accelerated progression of small adenomas, respectively.20,21
Against a backdrop of genomewide hypomethylation, a distinct subset of CRCs shows coordinate hypermethylation of many CpG-rich promoters, conferring the CpG island methylator phenotype (CIMP), with transcriptional attenuation of tumor suppressor genes such as HIC1 and Wnt-inhibiting SFRPs.22,23 Whole-genome methylation analyses have confirmed the existence of this once controversial entity24 and features that distinguish it from KRAS-mutant CIN disease—origin in sessile serrated adenomas; strong association with BRAF-mutant, right-sided MSI-hi tumors with MLH1 gene methylation;25 and distinctive gene expression patterns.26
Although the features cited previously, especially MMR and CIMP, overlap to some degree, CRCs seem to fall into three general categories: (1) traditional, (2) alternative, and (3) serrated, which are characterized by CIN, DNA mismatch repair, and CIMP, respectively (see Fig. 13.1). The Cancer Genome Atlas (TCGA) Network classified CRCs into nonhypermutated (generally CIN+) and hypermutated (encompassing MMR and CIMP) categories.26 Other variations on the classical adenoma–carcinoma sequence are also recognized. A 10-fold elevated risk of CRC in patients with long-standing ulcerative colitis (UC)27 likely reflects heightened mutation in a setting of ongoing mucosal injury and repair. UC-associated CRCs often arise within flat adenomatous plaques and nonadenomatous areas of dysplasia. Compared to sporadic cases, TP53 mutations occur earlier in the cancer sequence,28 APC inactivation is less frequent, and methylation of the p16INK4a tumor suppressor gene is more common.29
INHERITED SYNDROMES OF INCREASED CANCER RISK HIGHLIGHT EARLY EVENTS AND CRITICAL PATHWAYS IN COLORECTAL TUMORIGENESIS
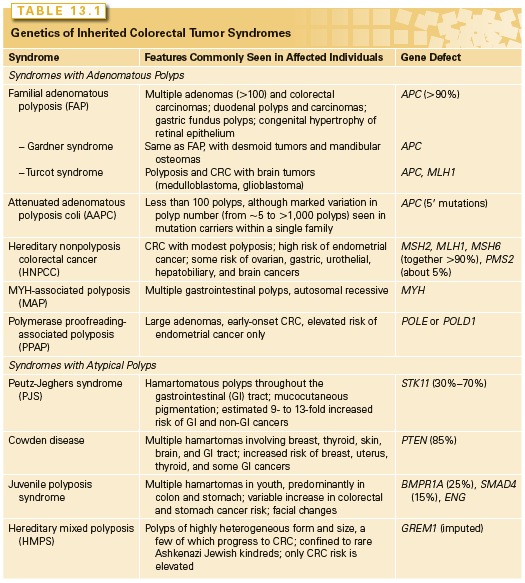
Two uncommon but highly penetrant Mendelian syndromes, familial adenomatous polyposis (FAP) and hereditary nonpolyposis colorectal cancer (HNPCC), together account for almost 5% of cases. MYH-associated polyposis (MAP), polymerase proofreading-associated polyposis (PPAP), familial juvenile polyposis (FJP), Peutz-Jeghers syndrome (PJS), and Cowden disease, each occurring in fewer than 1 in 200,000 births, also elevate the risk of CRCs (Table 13.1). Knowledge about the genes responsible for these inherited disorders allows for accurate molecular diagnosis, risk assessment, and targeted prevention in affected families. It also profoundly informs understanding of the considerably larger proportion of sporadic cases.
Familial Adenomatous Polyposis and the Central Importance of Wnt Signaling
FAP is an autosomal-dominant monogenic disorder that underlies about 0.5% of all CRCs. Individuals develop hundreds to thousands of colonic polyps by their early 20s, and the lifetime risk of CRC approaches 100% at a median age of 39. Extraintestinal manifestations include (1) duodenal and gastric adenomas; (2) congenital hypertrophy of the retinal pigmented epithelium; (3) osteomas and mesenteric desmoid tumors in the Gardner syndrome variant30; (4) brain tumors in the Turcot syndrome variant31; and rarely, (5) cutaneous cysts, thyroid tumors, or adrenal adenomas. Although most of these extraintestinal features are benign, rare patients develop hepatoblastoma or thyroid cancer. Reflecting similar homeostatic mechanisms in the small and large bowel epithelia, a 5% to 10% risk of periampullary adenocarcinoma mandates endoscopic monitoring of the duodenum after prophylactic colectomy.32
The gene responsible for FAP, adenomatous polyposis coli (APC), encodes a 300 kDa protein. Germ-line mutations occur throughout the locus, clustering in the 5′ half and exon 15,33 mostly introducing premature truncations. A few mutations correlate with phenotypic severity or specific extraintestinal manifestations, but identical mutations can produce different features. Mutations clustered in the extreme 5′ or 3′ ends of APC exons lead to the variant attenuated APC, with few polyps or CRCs developing late in life.34 The I1307K allele, present in Ashkenazi Jews, barely doubles the lifetime risk of CRC over the background and does not affect APC protein function but replaces an (A)3T(A)4 coding sequence with an (A)8 tract that is occasionally targeted for nearby truncating mutations.35 The identification of specific APC mutations in probands allows for a reliable testing of family members. Minimal recommendations for carriers include screening colonoscopy annually after age 10, gastroduodenoscopy after age 25, and treatment with nonsteroidal anti-inflammatory drugs to reduce CRC risk.36 A prophylactic colectomy is highly recommended, with continued vigilance over the rectal stump and other at-risk tissues.
The larger significance of the APC gene derives from its somatic inactivation in about 80% of sporadic CRCs and adenomas,37 including tiny polyps without dysplasia. APC inactivation is a rate-limiting step in the development of most polyps, and knowledge of its cellular functions supports this gatekeeper role. Attesting to the tumor suppressor function, CRCs that arise sporadically or in FAP show loss of heterozygosity and biallelic APC inactivation, with one copy usually lost by deletion. Because APC encodes several functional domains, truncated mutant proteins might interfere with various cellular activities. Disruption of its role in chromosome segregation might, for example, contribute to CIN.38 However, attention on APC rightly centers on its control of the Wnt signaling pathway. About half the sporadic CRCs with intact APC function carry activating point mutations in CTNNB1,39,40 which encodes β-catenin, a transcriptional effector of Wnt signaling; many of the rest carry fusions of genes that encode R-spondin cofactors.41 Moreover, acute APC loss in mice produces intestinal defects identical to those observed upon Wnt pathway activation.42 APC mutations are uncommon in sporadic cancers outside the intestine.
Wnts are secreted glycoprotein morphogens with diverse developmental and homeostatic functions, and, especially in the intestine, their role is intimately tied to that of R-spondins.43,44 In the absence of Wnt ligands, cells use a complex containing APC, AXIN2, and other cytosolic proteins to promote casein kinase I- and glycogen synthase kinase (GSK)-3β-mediated phosphorylation of conserved serine and threonine residues at the N-terminus of β-catenin; this phosphorylation targets β-catenin for ubiquitin-mediated proteasomal degradation. The binding of Wnt ligands to a surface complex containing a FRIZZLED protein and the obligate coreceptor LRP5/6 inhibits the APC-AXIN2 destruction complex and stabilizes a pool of cytosolic β-catenin (Fig. 13.2), distinct from the abundant cellular stores that attach to E-cadherin on the inner cell surface. Accumulated β-catenin translocates to the cell nucleus, where it coactivates genes bound by sequence-specific transcription factors of the T-cell factor (TCF) family. Of the four proteins in this family, TCF4 is especially important in normal bowel epithelium and CRCs39,45 and nuclear β-catenin is necessary to activate target genes.46
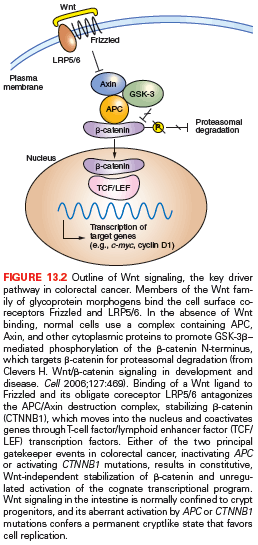
CTNNB1 mutations in CRC invariably target residues for N-terminal phosphorylation and the mutant protein hence, resists degradation. Thus, both alternative gatekeeper lesions in CRC, inactivating APC or activating CTNNB1 mutations, result in constitutive, Wnt ligand-independent stabilization of β-catenin. R-SPONDIN genes are translocated in up to 10% of cases, all with wild-type APC and CTNNB1,41 and the gene fusions are probably pathogenic because they potentiate Wnt signaling. TCF4 (also known as TCF7L2) mutations are surprisingly common,26 but their roles and effects are unclear, as is the functional significance of rare AXIN2 mutations in MSI+ cases47 and of TCF3 or TCF4 gene fusions in microsatellite stability (MSS) cases.48
Normal intestinal crypt stem and progenitor cells require Wnt signaling to proliferate, and APC or CTNNB1 mutation-induced loss of this ligand dependence liberates cells from a potent regulatory restraint. Accordingly, the Wnt-dependent transcriptional program in CRC cell lines overlaps significantly with that in intestinal crypts.49 Cycling crypt base cells that express the surface marker LGR5 are especially vulnerable to Wnt-induced transformation in mice, suggesting that CRC arises from stem cells and not from mature descendants.50 Laboratory evidence suggests that even advanced CRCs remain dependent on constitutive Wnt pathway activity.49 The TCF4–β-catenin complex controls hundreds of candidate genes,49,51 but the individual significance of most target genes is unclear. Apc mutant mice lacking CD44 and selected other Wnt-pathway targets develop fewer adenomas,52 but MYC seems especially vital because its absence in mouse intestines completely abrogates effects of acute APC loss.53,54
Hereditary Nonpolyposis Colorectal Cancer and the Role of DNA Mismatch Repair
HNPCC, or Lynch syndrome, an autosomal-dominant disorder that confers about 70% lifetime risk of developing CRCs, usually before age 50, is estimated to account for 2% to 4% of all cases. Affected individuals do not lack colonic polyps (nearly all CRCs, syndromic or sporadic, arise within benign precursors), but develop many fewer polyps than patients with FAP, a condition that must be excluded to satisfy diagnostic criteria for HNPCC (Table 13.2).55 Cancers tend to develop in the ascending colon and patients are also predisposed to develop tumors of the endometrium (35% to 50% lifetime risk), ovary and upper urothelium (7% to 8% risk), stomach, small intestine, biliary tract, and brain—a spectrum reflected in the revised Amsterdam II criteria (see Table 13.2).56 Cancers in HNPCC show pronounced variation in the lengths of microsatellite DNA sequences, and MSI at two or more among a panel of five mono- and dinucleotide tracts (BAT26, BAT25, D5S346, D2S123, and D17S250) confers the MSI-hi designation. CRCs harboring CIN represent a distinct class and show MSS or, in a small fraction, instability of unclear significance at just one of the five test regions (MSI-lo).

HNPCC results from germ-line mutations in any of several genes that enable DNA mismatch repair (MMR), a proofreading process that corrects base-pair mismatches and short insertions and deletions in the normal course of DNA replication. MMR in mammalian cells is mediated by homologs of bacterial and yeast repair proteins, MutS homologs (MSH) 1 through 6, MutL homologs (MLH) 1 through 3, PMS1, and PMS2. Both MLH1 and PMS2 are recruited to sites of DNA mismatch as a MutLα complex and in turn recruit MSH2-MSH6 (MutSβ) or MSH2-MSH3 (MutSβ) heterodimers to sites of 1-bp or 2- to 4-bp errors, respectively. These proteins efficiently excise the strand that carries the mismatch and resynthesize and ligate the repaired DNA. Germ-line mutations in MSH2, MLH1, MSH6, and PMS2 together explain about 95% of kindreds,57–59 including a subset with germ-line loss of the stop codon of TACSTD1, which results in silencing of its 3′ neighbor MSH2 by hypermethylation.60,61
MSI-hi colon cancers typically show exophytic growth, lymphoid infiltrates, mucinous signet-ring differentiation, and a medullary growth pattern; Bethesda guidelines (see Table 13.2) combine clinical and phenotypic features to facilitate a diagnosis of HNPCC.62 When these criteria are met, tumor DNA should be tested either for MSI in a simple, polymerase chain reaction (PCR)-based assay or by immunohistochemistry for the absence of the commonly implicated MLH1, MSH2, and MSH6 proteins.63 Because Bethesda guidelines might miss up to a quarter of cases, experts now recommend testing all colon cancers arising in patients under 70 years.64,65 Coupled with thorough personal and family histories, positive results should prompt DNA testing for MLH1, MSH2, MSH6, or PMS2 mutations.66 The identification of mutant alleles and carriers allows for the targeting of cost-effective screening and interventions that are proven to reduce mortality65 (e.g., preventive colonoscopy screening every 1 to 2 years, starting around 30 years of age; family counseling; aspirin therapy). Carriers should consider prophylactic subtotal colectomy, hysterectomy, and oophorectomy; females should at least have an annual endometrial evaluation soon after age 30.
In incipient cancers, random events first disrupt function of the wild-type allele of a mutant MMR gene and the resulting mutator phenotype promotes DNA replication errors at rates 102 to 103 times higher than the background.13,57 Consequently, adenomas progress into carcinomas over 3 to 5 years instead of 2 or more decades.67 Paradoxically, the prognosis for patients with MSI-hi CRCs is better than for those with sporadic MSS disease, perhaps because many resulting somatic mutations place tumors at a disadvantage. Both of the most commonly inactivated genes, ACVR2A and TGFBR2, encode receptors for specific ligands of the transforming growth factor beta (TGF-β) family and contain vulnerable mononucleotide tracts in their coding sequences.26,68 TGF-β inhibits the proliferation of intestinal epithelial cells, and biallelic TGFBR2 inactivation is detected in over 90% of MSI-hi and 15% of MSS, sporadic CRCs.69 Other genes mutated in familial MSI-hi colon tumors encode the proapoptotic genes CASP5 and BAX70; transcription factor genes, including TCF471; and the epidermal growth factor receptor (EGFR)72; but KRAS and, especially, BRAF mutations are rare. CRC pathogenesis requires deregulated APC–β-catenin gatekeeper function irrespective of MMR status.73
MSI-hi is observed in 12% to 15% of sporadic cases of CRC,74 often in older patients with early-stage disease. Such tumors seem to arise from sessile serrated adenomas in the ascending colon and do not reflect unrecognized germ-line mutation or somatic disruption of a known MMR gene.75 Rather, most but not all cases reflect MLH1 gene inactivation by biallelic promoter hypermethylation and show activating BRAF mutations and CIMP.76–78
MYH-Associated Polyposis and Polymerase Proofreading-Associated Polyposis
Germ-line mutations in MYH, a homolog of the Escherichia coli base excision-repair gene MutY cause MAP, a recessively inherited syndrome of multiple adenomas and CRC.79 CRC develops later in life than in FAP, polyp numbers vary widely, and extracolonic tumors are less frequent than in HNPCC (see Table 13.1). Because MYH is a DNA glycosylase that helps repair oxidative DNA damage, tumors are not associated with MSI but with somatic G:C to T:A mutations, including in the APC gene. Two alleles, Y165C and G382D, account for most cases, and CRCs develop in homozygotes or compound heterozygotes, with no risk elevation in monoallelic carriers.80 Surveillance recommendations are the same as in HNPCC.
Rare kindreds with a dominantly inherited high risk of CRC carry particular germ-line defects in POLE and POLD1, which encode the proofreading exonuclease activities of the leading- and lagging-strand DNA polymerases ε and δ, respectively.81 Clinical findings of large adenomas and early-onset CRCs resemble those in HNPCC or MAP, including elevated endometrial cancer risk in women with mutant POLD1. Tumors carry thousands of mutations, but stable microsatellite tracts reveal a third familial mutator mechanism in CRCs. Hypermutation in the absence of MSI accounts for about 3% of sporadic CRCs, nearly all of which have a somatic POLE exonuclease domain and also APC mutations.26 POLE and POLD1 are nonclassical tumor suppressors because, instead of deletion or truncation, specific missense mutations affect proofreading function, and the wild-type allele is usually retained. Until further characterization of PPAP leads to formal surveillance recommendations, it is prudent to adopt those developed for HNPCC.
Familial Juvenile Polyposis and the Peutz-Jeghers, Hereditary Mixed Polyposis, and Cowden Syndromes
Patients with FJP develop premalignant hamartomatous polyps in the stomach or the small or large intestine by adolescence82,83 and may give a revealing family history, although a significant minority represent the first case in their families. Germ-line mutations in genes encoding the bone morphogenetic protein (BMP) receptor BMPR1A, the accessory TGF-β receptor endoglin, ENG, or the SMAD4 signal transducer emphasize the role of TGF-β signaling in disease pathogenesis (see Table 13.1).84,85 Indeed, sporadic CRCs are often insensitive to the growth inhibitory effects of TGF-β and loss of BMP function in mice expands stem and progenitor cells, leading to polyposis or ectopic crypts.86–88 Not all patients carry these mutations, indicating that additional genes remain undiscovered. Notably, a conditional Smad4 deletion from mouse intestinal cells does not affect growth, whereas selective loss in T lymphocytes causes intestinal mucosal thickening and polyps.89 These findings confound simple interpretations of TGF-β function and implicate stromal inflammation in intestinal tumorigenesis.
Patients with PJS also develop benign tumors that contain differentiated but disorganized cells (hamartomas), mainly in the small intestine but also in the colon or stomach, sometimes leading to hemorrhage or intussusception. PJS shows autosomal-dominant inheritance and is associated with macular lesions on the skin and buccal mucosa; bladder and bronchial polyps; and a propensity to develop a range of cancers, including those of the lung, breast, and female reproductive organs (see Table 13.1). The lifetime risk for all cancers exceeds 90%, the incidence of small intestine, stomach, and pancreas cancers is 50 to 500 times higher than the general population, and CRC risk is elevated nearly 100-fold.90 Serine–threonine kinase 11 (STK11, also known as LKB1), the implicated tumor suppressor gene,91 acts at the nexus of diverse cellular pathways and functions. Its principal activity, exerted through the adenosine monophosphate (AMP)-activated protein kinase AMPK, seems to be in linking nutrient and energy utilization to controls over cellular structure, particularly cell polarity.92 STK11 also modulates the Rheb-GDP:Rheb-GTP cycle and downstream activities of the tuberous sclerosis gene TSC2 and the mammalian target of rapamycin (mTOR),93 key regulators of protein synthesis and cell growth. The roles of STK11 in cell polarity and metabolism are busy areas of research, likely to hold vital clues into CRC pathogenesis and rational therapy.
Confined to rare Ashkenazi Jewish kindreds descended from a single founder, patients with HMPS develop polyps of several diverse morphologies and CRCs but no other cancers.94 A 40-kb duplication upstream of GREM1 seems to activate a distant enhancer of this BMP antagonist gene, driving ectopic and high expression in the colonic epithelium.95 The resulting increased crypt cell turnover likely accelerates the accumulation of oncogenic mutations. GREM1 is also the locus of a CRC risk allele identified at 15q13.3 in a GWA study.96
Cowden disease encompasses diverse mucosal lesions, specific cutaneous lesions (facial trichilemmomas and acral verrucous papules), and breast fibroadenomas, neurofibromas, lipomas, and meningiomas.97 The syndrome results from germ-line mutations in PTEN, a tumor suppressor gene encoding the phosphatase and tensin homolog deleted on chromosome 10,98 and the second most frequently mutated gene in cancers after TP53. The lipid phosphatase PTEN dephosphorylates key phosphoinositide (PI) signaling molecules99 and accordingly regulates intracellular growth signaling negatively through PI-3 kinase and its downstream effectors AKT and mTOR. The CRC risk in Cowden syndrome is modest, and PTEN mutations are rare in sporadic cases, but protein immunostaining is lost in about 40% of CRCs, often as a result of promoter hypermethylation,100 thus highlighting its tumor suppressor role.
Significance of Inherited Syndromes of Elevated Colorectal Cancer Risk
Following a clinical diagnosis of the previous Mendelian syndromes, patients and family members should be tested for pertinent germ-line mutations, receive genetic counseling, and enter programs for cancer prevention and screening. The corresponding molecular defects profoundly inform an understanding of sporadic CRCs, in particular, revealing the seminal role of Wnt signaling and early, rate-limiting effects of APC inactivation or CTNNB1 activation. Similarly, STK11 and PTEN loss in inherited and sporadic CRCs shed light on crucial molecular pathways, whereas HNPCC and PPAP help classify the disease and reveal the significance of features such as MSI, which is present in 12% to 15% of sporadic cases. Even in the absence of a recognized predisposition syndrome, individuals with a history of CRC in a first-degree relative are up to 4 times more likely to develop CRC than those without a family history. Specific environmental factors that compound the risk of developing CRCs are complex and insufficiently characterized, but include obesity, excessive consumption of red meat, physical inactivity, and vitamin D deficiency.101 Because many of these factors converge on insulin signaling, some experts propose that insulin and insulinlike growth factors play a seminal role in CRCs.102 However, three of every four CRCs arise in individuals lacking a well-defined risk factor, and it is unknown to what extent particular genotypes confer sensitivity to environmental variables.
The cancer spectrum in HNPCC or PPAP and the particular predilection for CRC remain unexplained. Colonic, endometrial, and selected other epithelia may be especially sensitive to the kind of mutations that occur in the setting of defective DNA mismatch and base-excision repair, loss of wild-type tumor suppressor alleles may occur more readily in these tissues, or they may lack repair safeguards that protect other cell types.
Insights from Genomewide Association Studies
The quarter or more of sporadic CRC cases with a familial component103,104
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


