Based on gene expression profiling, GBMs cluster into four groups: classical (chromosome 7 amplification with chromosome 10 loss, deletion of CDKN2A), mesenchymal (NF1 focal deletions or mutations), proneural (IDH1 mutant or PDGFRA amplification), and neural (EGFR amplification with a neural expression signature).23 These classifications have a utility for predicting response to therapy; for example, the classical subtype responds to more intensive therapy whereas the proneural subtype shows no benefit to this regimen.23 Another prognostic indicator is the CpG island methylator phenotype (G-CIMP) demonstrated in IDH1 mutant proneural tumors, which had a significantly better survival (median of 150 weeks) compared to proneural G-CIMP–negative patients (median survival of 42 weeks) or other GBM subtypes (median survival 54 weeks).6 Also clinically useful to predict response to therapy is O-6-methylguanine-DNA methyltransferase (MGMT) promoter methylation status,19,26 because tumors with silenced MGMT are unable to remove the alkyl groups deposited on the O6 position of guanine by alkylating agents such as temozolomide.27
Anaplastic (grade III) gliomas, including anaplastic astrocytomas, anaplastic oligoastrocytomas, and anaplastic oligodendrogliomas, are not as well characterized genomically as GBMs. Clinically, they can arise without a prior history of low-grade glioma (presumed de novo) or through progression from low-grade gliomas (secondary anaplastic glioma). Grade III gliomas have a high risk of progression to GBM, although the rate of progression and prognosis varies by histology, with anaplastic astrocytomas having a 5-year overall survival of 26.5% and anaplastic oligodendroglioma having a 5-year overall survival of 50.7%.1 IDH1 mutations have been observed in 75% to 90% of grade III gliomas.4,16,28 Anaplastic astrocytomas, accounting for 1.7% of primary brain tumors,1 commonly have mutations in ATRX, IDH1, and loss of p53 as well as alterations to the Rb pathway (including RB1 loss, CDKN2A deletion, and CDK4/6 amplification).16,18,28 It is believed that additional hits in the RTK/Ras/PI3K pathway, including LOH at chr10q, lead to progression to frank glioblastoma.17,18,21 Anaplastic oligodendrogliomas are rare (0.5% of primary brain tumors),1 and the progression from grade II oligodendrogliomas (characterized by chr1p/19q loss and IDH1-CIC/FUBP1 mutations) is likely mediated by additional deletion of CDKN2A and PTEN.13 Like GBMs, recurrent TERT promoter mutations (C288T or C250T) have been identified in anaplastic gliomas, reported in 14.8% of anaplastic astrocytomas, 26.7% of anaplastic oligoastrocytomas, and 88.4% of anaplastic oligodendrogliomas.25 The differences in TERT promoter mutations between the subtypes is probably due to the higher frequency of ATRX mutations in anaplastic astrocytomas, because TERT promoter mutations are mutually exclusive with ATRX mutations in GBMs and other tumors.25
Pediatric Low-Grade Gliomas
Pediatric low-grade gliomas (WHO grade I and II) are the most common brain tumors in children1 and can be broadly divided into nondiffuse (e.g., pilocytic astrocytomas) and diffuse gliomas. Pilocytic astrocytomas (WHO grade I) are histologically benign tumors with a low probability of malignant progression that are usually pediatric, are primarily found in the cerebellar hemisphere (67%)10, and are cystic.2 The mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway was originally implicated in driving the formation of pilocytic astrocytomas through studies of the hereditary tumor syndrome neurofibromatosis type 1, in which ~15% of patients with germline loss-of-function mutations in NF1, a negative regulator of Ras signaling, develop pilocytic astrocytomas in addition to café-au-lait spots and cutaneous neurofibromas.29 Further investigation has revealed that MAPK/ERK pathway activation is crucial to sporadic pilocytic astrocytoma formation, with 90% of cerebellar pilocytic astrocytomas demonstrating a KIAA1549-BRAF (B-Raf proto-oncogene, serine/threonine kinase) fusion gene that results in constitutively active BRAF signaling via a truncation of the BRAF autoinhibitory domain.30–33 Pilocytic astrocytomas can also have constitutive activation of MAPK/ERK through somatic BRAF V600E mutations, Kirsten rat sarcoma viral oncogene homolog (KRAS) mutations, Raf-1 proto-oncogene, serine/threonine kinase (RAF1) fusions, and NF1 loss-of-function mutations.32,33 Approximately 20% of noncerebellar pilocytic astrocytomas lack the KIAA1549-BRAF fusion, and these fusion-negative tumors have recently been discovered to activate MAPK signaling through additional methods, including fibroblast growth factor receptor 1 (FGFR1) alterations (mutations, tyrosine kinase domain [TK] duplications, and gene fusions with transforming, acidic coiled-coil containing protein 1 [TACC1]),32,33 neurotrophic tyrosine kinase, receptor, type 2 (NTRK2) fusions resulting in TK truncations that are predicted to induce constitutive dimerization,33 and protein tyrosine phosphatase, non-receptor type 11 (PTPN11) hotspot mutations in tumors co-mutated for FGFR1.33
Pediatric diffuse gliomas, including diffuse astrocytomas, gangliogliomas, angiocentric gliomas, pleomorphic xanthoastrocytomas, oligodendrogliomas, and oligoastrocytomas,32,34 differ from pilocytic astrocytomas in their diffuse growth pattern, anatomic location (generally supratentorial), and their propensity for malignant transformation.2 In a recent study by Zhang et al.,32 52% (12 out of 23) of diffuse astrocytomas (WHO grade II) demonstrated ERK/MAPK signaling activation via FGFR1/3 alterations (fusions or FGFR1 TK duplication), BRAF alterations (V600E mutations or fusions), or KRAS activating mutation (Q61H). This same series identified common, recurrent FGFR1 alterations in oligodendrogliomas (3 out of 5: FGFR1 TK duplications) and oligoastrocytomas (6 out of 8, 4 FGFR1 TK duplications; 1 fusion; 1 mutation).32 A subset of ERK/MAPK activated diffuse astrocytomas (2 out of 12; 16.67%) had the recurrent H3 histone, family 3A (H3F3A) K27M mutation, an alteration that has been frequently described in pediatric glioblastomas.32
The majority of other diffuse low-grade gliomas also demonstrate MAPK/ERK activation. Pleomorphic xanthoastrocytomas (WHO grade II) are rare, supratentorial astrocytomas that are found in the pediatric population two-thirds of the time.35 The 5-year overall survival is 81%, and malignant progression can occur but is generally rare.36 A recent study showed that 7 out of 10 pleomorphic xanthoastrocytomas had BRAF V600E mutations.32 Similarly, this study found 55.6% (5 out of 9) of gangliogliomas showed ERK/MAPK activation by BRAF alterations (V600E = 3; BRAF fusion production = 2).32
In contrast to pilocytic astrocytomas, about one-quarter of pediatric diffuse astrocytomas (6 out of 23; 26%) demonstrated alterations in v-myb avian myeloblastosis viral oncogene homolog (MYB), including gene fusion with protocadherin gamma subfamily A, 1 (PCDHGA1) or episome formation, and one case of v-myb avian myeloblastosis viral oncogene homolog-like 1 (MYBL1) rearrangement.32 In an independent, copy number alteration study, Ramkissoon et al.34 identified focal amplification resulting in tandem duplication/truncation of MYBL1 in 28% (5 out of 18) of pediatric diffuse astrocytomas. These MYB/MYBL1 alterations were not observed in pilocytic astrocytomas, although two out of two angiocentric gliomas from Zhang et al.32 and an additional two angiocentric gliomas from Ramkissoon et al.34 bore MYB or MYBL1 fusions, providing further implication of this pathway as specific to a subset of diffuse pediatric gliomas.
Pediatric High-Grade Gliomas
Although histologically similar to adult glioblastomas, a subset of pediatric glioblastomas have distinct genomic hits driving tumor formation that implicate epigenetic dysregulation in the formation of these tumors. In a study by Schwartzentruber et al.,37 recurrent mutations in histone variant H3.3 (H3F3A K27M; G34R/V), commonly co-occurring with damaging mutations in the chromatin remodelers ATRX or death-domain associated protein (DAXX) and TP53, drove pediatric glioblastoma formation in 31% of pediatric GBMs. This percentage may be higher in diffuse intrinsic pontine gliomas (DIPG), where an independent study found that 78% of DIPG contained somatic K27M mutations in H3F3A or histone cluster 1, H3b (HIST1H3B), whereas H3F3A G34R was restricted to non–brain stem pediatric glioblastomas.38 Recently, H3K27 K27M was characterized to be a dominant-negative mutation that results in a genome-wide reduction of H3K27me3 repressive marks due to altered binding with the polycomb repressive complex 2 (PRC2) and also causes global DNA hypomethylation.39 In pediatric glioblastomas, these H3.3 mutations were mutually exclusive with the neomorphic IDH1 R132H,8,37 which was rarely seen in pediatric GBM but has been shown to cause epigenomic dysregulation and DNA hypermethylation (G-CIMP) phenotype.9 Similar to pediatric low-grade gliomas, the BRAF V600E mutation has been reported in ~10% of pediatric high-grade gliomas40; however, unlike the pediatric low-grade gliomas, this mutation commonly co-occurs with homozygous CDKN2A/B deletion.41
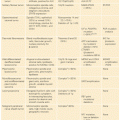
In terms of structural variation, pediatric glioblastomas differ from the classic, primary adult GBM findings of chr7 amplification (74% in adult versus 13% in pediatric) and chr10 loss (80% adult versus 35% pediatric).42 However, pediatric glioblastomas have more frequent 1q gain (30% in pediatric versus 9% in adult), frequent focal amplification of PDGFRA in 12% of tumors, and frequent focal, homozygous deletion of CDKN2A/B in 19% of tumors.42 The driver events for adult and pediatric gliomas are summarized in Table 22.1.
MENINGIOMAS
Meningiomas are the most common primary intracranial tumor, accounting for one-third of all primary brain tumors.1 Thought to arise from the arachnoid layer of the meninges, meningiomas can be found throughout the neuraxis, and are notable for a wide variety of histologic subtypes (15 according to the 2007 WHO classifications).43 Although 70% to 80% of meningiomas are benign (WHO grade I), the remaining subset can exhibit more aggressive behavior (WHO grade II and grade III).43 Recent genomic studies have revealed that 80% of meningiomas can be neatly categorized into three clinically relevant, mutually exclusive genetic groups with differences in histology, anatomic location, and likelihood for malignant progression (Fig. 22.2).44–46
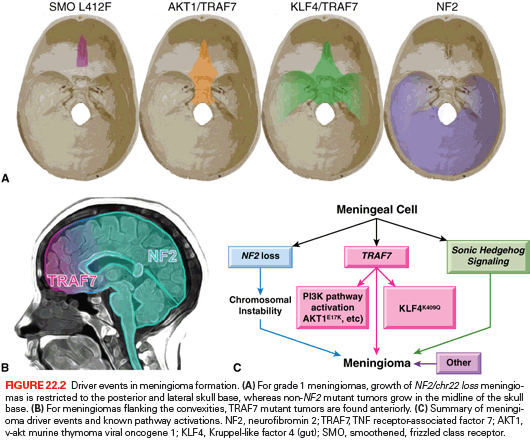
The first and largest group, NF2/chr22 loss tumors, is characterized by biallelic loss of the tumor suppressor neurofibromin 2 (merlin, NF2). One of the hallmarks of the inherited tumor syndrome neurofibromatosis type II is multiple meningiomas due to germline mutations in the tumor suppressor NF2,47 and biallelic loss of NF2 drives ~50% of sporadic meningioma formation48 and is a risk factor for malignant transformation. At least 75% of WHO grade II tumors have NF2/chr22 loss.43,44 NF2/chr22 loss tumors are more likely to form in the meninges flanking the cerebral convexities, but when they grow in the skull base, the growth is restricted to the posterior and lateral portions of the skull.44 NF2/chr22 loss is also commonly found in spinal meningiomas. Rarely, NF2/chr22 loss
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


