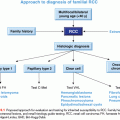
HEREDITARY BREAST CANCER
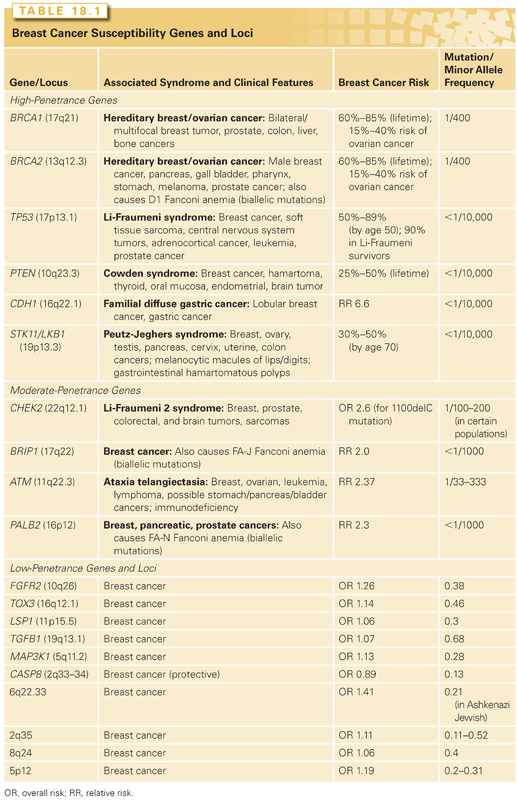
One of the most important risk factors for breast cancer is family history. Although familial forms comprise nearly 20% of all breast cancers, most of the genes responsible for familial breast cancer have yet to be identified. Breast cancer susceptibility genes can be categorized into three classes according to their frequency and level of risk they confer: rare high-penetrance genes, rare intermediate-penetrance genes, and common low-penetrance genes and loci (Table 18.1).4
High-Penetrance, Low-Frequency Breast Cancer Predisposition Genes
BRCA1 and BRCA2
BRCA1 and BRCA2 mutations account for approximately half of all dominantly inherited hereditary breast cancers. These mutations confer a relative risk of breast cancer 10 to 30 times that of women in the general population, resulting in a nearly 85% lifetime risk of breast cancer development.5 BRCA1 and BRCA2 mutation carriers are quite rare among the general population; however, the prevalence is substantially higher in certain founder populations, most notably in the Ashkenazi Jewish population, where the carrier frequency is 1 in 40.
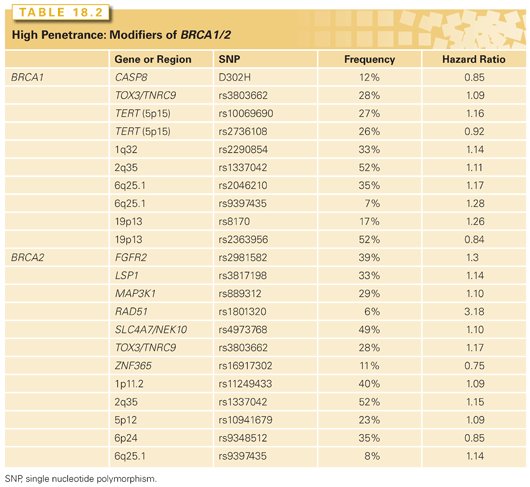
More than a thousand germ-line mutations have been identified in BRCA1 and BRCA2. Pathogenic mutations most often result in truncated protein products, although mutations that interfere with protein function also exist.4,5 Interestingly, the penetrance of pathogenic BRCA1 and BRCA2 mutations and age of cancer onset appear to vary both within and among family members. Specific BRCA mutations as well as gene–gene and gene–environment interactions as potential modifiers of BRCA-related cancer risk are areas of active investigation.6,7 Variation in risk may be explained by genetic modifiers that are different for both BRCA1 and BRCA2 mutation carriers. These alleles have been primarily identified from studies of the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA).8 The commonly identified single-nucleotide polymorphisms (SNP) that modify BRCA1/2 are listed in Table 18.2 with their gene location, associated risks, and frequency. Evidence from published studies shows that these modifying SNPs combine multiplicatively, and therefore, may significantly alter a mutation carrier’s risk depending on the number of risk alleles present.9,10 In addition to retrospective studies, more recently, common breast cancer susceptibility alleles were evaluated prospectively to assess cancer risk conferred among unaffected BRCA1/2 mutation carriers.11 A risk score was constructed and divided into tertiles for seven BRCA2-associated variants and four BRCA1-associated variants. Among BRCA2 mutation carriers, breast cancer risks were significantly different when stratified by tertiles; women in the highest tertile had a breast cancer risk of 72% by age 70 compared to 20% for those in the lowest tertile. There was no significant difference by risk score for BRCA1 mutation carriers.
BRCA1-related breast cancers are characterized by features that distinguish them from both BRCA2-related and sporadic breast cancers.4 BRCA1-related tumors typically occur in younger women and have more aggressive features, with high histologic grade, high proliferative rate, aneuploidy, and absence of estrogen and progesterone receptors and human epidermal growth factor receptor 2 (HER2). This triple-negative phenotype of BRCA1-related breast cancers is further characterized by a basallike gene expression profile of cytokeratins 5/6, 14, and 17, epidermal growth factor, and P-cadherin.12 Although BRCA1 and BRCA2 genes encode large proteins with multiple functions, they primarily act as classic tumor suppressor genes that function to maintain genomic stability by facilitating double-strand DNA repair through homologous recombination.12,13 When loss of heterozygosity (LOH) occurs via loss, mutation, or silencing of the wild-type BRCA1 or BRCA2 allele, the resultant defective DNA repair leads to rapid acquisition of additional mutations, particularly during DNA replication, and ultimately sets the stage for cancer development.
The integral role of BRCA1 and BRCA2 in double-strand DNA repair holds potential as a therapeutic target for BRCA-related breast cancers. For example, platinum agents cause interstrand cross-links, thereby blocking DNA replication and leading to stalled replication forks. Poly (adenosine diphosphate [ADP]-ribose) polymerase-1 (PARP1) inhibitors additionally show promise as specific therapy for BRCA-related tumors. PARP1 is a cellular enzyme that functions in single-strand DNA repair through base excision and represents a major alternative DNA repair pathway in the cell.14,15 When PARP inhibition is applied to a background deficient in double-strand DNA repair, as is the case in BRCA-related tumor cells, the cells are left without adequate DNA repair mechanisms and ultimately undergo cell cycle arrest, chromosome instability, and cell death.4 Given their phenotypic similarities to BRCA1-related breast cancers, sporadic basallike breast tumors may display sensitivity to PARP inhibition as well.15 Phase II studies are currently under way to explore the use of PARP inhibitors in both BRCA– and basallike, non–BRCA-related breast tumors. There is much that remains to be understood about the optimal use of PARP inhibitors. Current challenges include, but are not limited to, identifying robust predictive biomarkers of response that can guide patient selection and understanding variations among PARP inhibitors in clinical development. Differences in potency and the mechanism of action have been well elucidated in recent preclinical studies,16–20 and the results of ongoing clinical trials will need to be interpreted in this context. Additionally, recent studies have also identified mechanisms of resistance to PARP inhibitors. One of these important mechanisms includes secondary mutations in the BRCA1/2 gene that can restore the open reading frame, and therefore, DNA repair functional activity,21–23 which renders tumors resistant to PARP inhibitors. Secondly, loss of tumor protein p53 binding protein 1 (TP53BP1) in BRCA1-deficient cells can restore DNA repair activity,24,25 and this may confer resistance.
Other High-Penetrance Genes
A small number of other high-risk, low-frequency breast cancer susceptibility genes exist, and they include TP53, PTEN, STK11/LKB1, and CDH1.4–6 These high-penetrance genes confer an eight- to tenfold increase in the risk of breast cancer as compared to noncarriers, but they collectively account for less than 1% of breast cancer cases. Like BRCA1 and BRCA2, these genes are inherited in an autosomal-dominant fashion and function as tumor suppressors.26 The hereditary cancer syndromes associated with each gene are usually characterized by multiple cancers in addition to breast cancer, as summarized in Table 18.1.
Moderate-Penetrance, Low-Frequency Breast Cancer Predisposition Genes
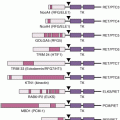
Four genes have been identified that confer an elevated but moderate risk of developing breast cancer, namely CHEK2, ATM, BRIP1, and PALB2 (see Table 18.1). Each of these genes confers approximately a two- to threefold relative risk of breast cancer in mutation carriers, although this risk may be higher in select clinical settings.5 Mutation frequencies in the general population are rare, on the order of 0.1% to 1%, although some founder mutations have been identified. Together, these genes account for approximately 2.3% of inherited breast cancer. The moderate relative risk of breast cancer of these genes in conjunction with the low population frequency renders this class of genes very difficult to detect with typical association studies. However, these genes were specifically selected for study as candidate breast cancer genes based on their known roles in signal transduction and DNA repair in close association with BRCA1 and BRCA2.6
Low-Penetrance, High-Frequency Breast Cancer Predisposition Genes and Loci
Both candidate gene and genome-wide association studies (GWAS) have identified a low-risk panel of approximately 10 different alleles and loci in 15% to 40% of women with breast cancer (see Table 18.1).5 Despite their frequency, the relative risk of breast cancer conferred by any one of these genetic variants alone is minimal, on the order of less than 1.5.4 Nevertheless, these alleles and loci may become clinically relevant in their suggestion of interactions with other high-, moderate-, and low-risk genes; these additive or multiplicative relationships could account for a measurable fraction of population risk. For example, association studies of fibroblast growth factor receptor 2 (FGFR2) and mitogen-activated protein kinase kinase kinase 1 (MAP3K1) within BRCA families showed that these SNPs conferred an increased risk in the presence of BRCA2 mutations.
Microsatellite Instability in Breast Cancer
There is emerging data that Lynch syndrome, an autosomal-dominant inherited disorder of cancer susceptibility caused by germ-line mutations in the DNA mismatch repair (MMR) genes including, MLH1, MSH2, MSH5, and PMS2, may increase the risk of breast cancer.27 Mutation carriers are at increased risk of colorectal and other cancers, but its association with breast cancer risk has been controversial. A prospective cohort study using the Colon Cancer Family Registry evaluated cancer risks among unaffected carriers and noncarriers with a pathogenic MMR gene mutation; notably, breast cancer risk was estimated to be fourfold among mutation carriers compared to the general population.27 A systematic review of breast cancer risk studies for Lynch syndrome mutation carriers showed mixed results; 13 studies did not observe an increased risk, whereas 8 studies observed an increased risk of breast cancer ranging from 2- to 18-fold compared to the general population.28 Further studies are needed to determine more precise estimates of breast cancer risk in Lynch syndrome carriers with longer follow-ups. These studies may also guide future breast cancer screening guidelines for this population.
MicroRNA and Cancer Susceptibility
Recent studies suggest that microRNA (miRNA) SNPs may also contribute to breast cancer susceptibility, and miRNAs appear to regulate many tumor suppressor genes and oncogenes via degradation of target miRNAs or repression of their translation. Thus, genetic variations in miRNA genes or miRNA binding sites could affect the expression of tumor suppressor genes or oncogenes and, thereby, affect cancer risk. For example, specific SNPs located within pre-mir-27a and mir-196a-2 genes have been associated with reduced breast cancer risk, which has been confirmed in a recent meta-analysis.29,30
SOMATIC CHANGES IN BREAST CANCER
The vast majority of breast cancers are sporadic in origin, ultimately caused by an accumulation of numerous somatic genetic alterations.1 Recent data suggest that a typical individual breast cancer harbors anywhere from 50 to 80 different somatic mutations.2 Many of these mutations occur as a result of erroneous DNA replication; others may occur through exposure to exogenous and endogenous mutagens. To date, hundreds of candidate somatic breast cancer genes have been identified through GWAS.31,32
Determining the role of each identified mutation in the development of breast cancer remains a substantial challenge. Data suggest that the vast majority of identified somatic DNA mutations in a given tumor are passenger mutations, representing harmless, biologically neutral changes that do not contribute to oncogenesis.1,2 Conversely, driver mutations confer a growth advantage on the cell in which they occur and appear to be implicated in cancer development. By definition, driver mutations are found in candidate cancer genes (CAN).32 A comprehensive catalog of somatic mutations and CAN genes has been accumulated through multiple studies. When specific driver mutations are cataloged among several different breast tumors, a bimodal cancer genomic landscape appears, comprising a small number of commonly mutated gene mountains among hundreds of infrequently mutated gene hills.1,2 Gene mountains correspond to the most frequently mutated genes found within breast tumors, such as TP53, CDH1, phosphatidylinositol 3-kinase (PI3K), cyclin D, PTEN, and AKT.6 Each individual gene hill, on the other hand, is typically found in less than 5% of breast tumors.1,33 This substantial heterogeneity of DNA mutations among breast tumors may explain the wide variations in phenotypes, both in terms of tumor behavior as well as responsiveness to therapy.
Historically, the focus of genetic research has been on the gene mountains, in part because they were the only mutations that available technology could identify. However, emerging data suggest that it is actually the gene hills that play a much more pivotal role in breast cancer, which is consistent with the idea that having a large number of mutations, each associated with a small survival advantage, drives tumor progression. Recent studies have shown that a substantial number of these infrequent somatic mutations sort out among a much smaller number of biologic groups and cell signaling pathways that are known to be pathogenic in breast cancer, thereby vastly reducing the complexity of the genomic landscape. Examples of such pathways include interferon signaling, cell cycle checkpoint, BRCA1/2-related DNA repair, p53, AKT, transforming growth factor β (TGF-β) signaling, Notch, epidermal growth factor receptor (EGFR), FGF, ERBB2, RAS, and PI3K. In short, it appears that common pathways, rather than individual gene mutations, govern the course of breast cancer development.33
Although recurrent point mutations are less common in breast cancer than other solid tumors, emerging data show that particular regions of the genome are commonly amplified, and these regions contain genes that drive cancer progression. The best example of an important amplified region is the 17q12 amplicon that harbors the HER2 oncogene. This amplicon leads to a more aggressive tumor phenotype, now the target of a highly successful antibody therapy, trastuzumab (Herceptin). It has been observed that RNA-mediated interference (RNAi) knockdown of coamplified genes within the 17q12 amplicon results in decreased cell proliferation and increased apoptosis.34 Thus, the 17q12 amplicon appears to encode a concerted genetic program that contributes to the oncogenesis.
There are several other amplicons, in addition to 17q12 (HER2), that seem to drive the cancer phenotype and have prognostic significance in breast cancers, for example, 11q13 (CCDN1) and 8q24 (MYC), 20q13.35 These regions contain gene sets that are important in DNA metabolism and in the maintenance of chromosomal integrity, suggesting that a response to DNA damaging agents used as anticancer therapy might be modulated by the presence of particular amplicons. Indeed, these coamplicons are frequent in HER2-amplified tumors and may modify tumor behavior and patient outcome.36,37 The contribution of these genomic alterations to functional consequences may lie not in the overexpression of individual genes, but of gene cassettes on the amplicon.
Direct clinical translation of the growing catalog of somatic alterations in breast cancer has yet to evolve. However, with advancing technology and further identification and categorization of genetic mutations, new opportunities for individualized diagnosis and treatment options are likely to emerge.
TRANSCRIPTIONAL PROFILING OF BREAST CANCER
The cellular programs that are encoded by DNA are enacted by transcription into messenger RNA (mRNA) and translated into protein. Not surprisingly, the DNA alterations described previously lead to either under- or overexpression of their associated mRNAs; consequently, abnormal gene expression patterns are a common finding in breast tumors. Gene expression profiling has been introduced into the clinical literature during the past decade because research suggests that assessing the expression of multiple genes in a tumor sample may reflect programs turned on by DNA alternations and predict tumor behavior. So-called molecular signatures hold promise for improving the diagnosis, the prediction of recurrence, and the selection of therapies for individual patients.
Several technologies have been developed to generate molecular signatures, including cDNA and oligonucleotide arrays and multiplex polymerase chain reaction (PCR) technologies. These technologies and newly developed statistical methodologies now allow for evaluations of hundreds and even thousands of mRNAs simultaneously with groupings of samples based on coexpressedgenes.
Molecular Classification of Breast Cancer
The seminal work by Perou et al.38 and Sorlie et al.39 suggests a classification of breast cancer subtypes based on gene expression patterns they termed molecular portraits of breast cancer. Among the categories they defined were the luminal A and B tumor types (typically estrogen-receptor [ER] or progesterone-receptor [PR] positive), HER2 gene-amplified tumors, and a class termed basal-like due to the expression of basal keratins. Recent large scale efforts by The Cancer Genome Atlas Network (TCGA)40 and the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC)41 groups have confirmed these earlier findings in addition to providing more detailed molecular portraits.
Luminal Subtypes
The luminal subtypes comprise the majority of breast cancers and are characterized by the expression of genes that are normally expressed in the luminal epithelial of the breast such as cytokeratins 8 and 18 and the luminal expression signature (ESR1, GATA3, FOXA1, XPB1, and MYB). Luminal subtypes comprise the majority of clinical ER-positive breast cancers and can be divided into two subgroups: luminal A and luminal B. Luminal A tumors are more common and are characterized by high expression levels of ER-related genes and low expression of the HER2 cluster and proliferation-associated genes. In contrast, luminal B tumors are characterized by lower expression levels of ER-related genes, variable expression of the HER2 cluster, and higher levels of proliferation-associated genes. Luminal A tumors have an overall better prognosis than luminal B tumors.
HER2-Enriched Subtype
The HER2-enriched subtype comprises approximately 10% to 15% of all breast cancers and overexpresses both HER2 and proliferation-associated genes and has lower expression of ER-related genes. Interestingly, more recent work by the TCGA demonstrates that not all cancers that are clinically HER2-positive as defined by an immunohistochemical (IHC) analysis and/or fluorescent in situ hybridization (FISH) fall into the HER2-enriched molecular subtype and vice versa. The majority of clinically HER2-positive breast cancers that are not considered part of the HER2-enriched subgroup by gene expression profiling fall into the luminal intrinsic subtype with overexpression of HER2.
Estrogen Receptor–Negative Subtypes
The ER-negative subtypes comprise a heterogeneous group of tumors that clinically are termed triple negative breast cancer (TNBC) because they typically lack ER, PR, and HER2, and are often referred to as triple negative, although not all basallike tumors are triple negative and visa versa. The basallike category of the ER-negative subsets were first identified with first-generation microarray technology and show a high expression of proliferation genes and basal cytokeratins5,31,34 and a loss of genes associated with cell cycle control, which confer an overall poor prognosis. Though basallike tumors are the most common of the ER-negative subtypes (50% to 75% of all ER-negative tumors) and comprise 15% to 20% of all types of breast cancer, other ER-negative subtypes also exist, which include the recently described claudin-low group, as well as interferon-rich, androgen receptor, normallike groups. Though the claudin-low subgroup has some similarities to basallike breast cancer, it is distinct because these tumors have low expression of the claudin genes that are involved in epithelial cell tight-tight junctions. The claudin-low tumors have been of particular interest because they posses stem cell–like features of epithelial–mesenchymal transition (EMT).42 To further characterize the heterogeneity of TNBC, a recent study did a clustering analysis of gene expression profiling of primary tumors and identified six distinct subtypes.43
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


