Classic as well as more recent large-scale genomic analyses have uncovered multiple genes and pathways important for bladder cancer development. Genes involved in cell-cycle control, chromatin regulation, and receptor tyrosine and PI3 kinase-mammalian target of rapamycin signaling pathways are commonly mutated in muscle-invasive bladder cancer. Expression-based analyses have identified distinct types of bladder cancer that are similar to subsets of breast cancer, and have prognostic and therapeutic significance. These observations are leading to novel therapeutic approaches in bladder cancer, providing optimism for therapeutic progress.
Key points
- •
There is a high rate of both mutations and genomic amplifications and deletions in muscle-invasive bladder cancer.
- •
FGFR3 is commonly activated in bladder cancer, through either mutation, gene fusion events, or elevated expression, and is a potential therapeutic target.
- •
Chromatin regulatory gene mutations, Cell-cycle gene mutations, amplifications (cyclins), and deletions (cyclin-dependent kinase inhibitors) are very common in bladder cancer.
- •
Viral infection seems to contribute to bladder cancer development in 5% to 10% of cancers.
- •
The main RNA expression subtypes of bladder cancer are basal and luminal, similar to breast cancer, and confer both prognostic and therapeutic significance.
- •
The following genes and pathways are frequently affected: the ERBB family (including EGFR, ERBB2, and ERBB3), FGFR3, and the PI3K-mTOR (PI3KCA, PTEN, and TSC1) signaling cascade, many of which are potentially targetable using small molecule kinase inhibitors or antibody therapeutics.
Background
Bladder cancer is a leading cause of morbidity and mortality, with nearly 400,000 new cases and 150,000 deaths worldwide. However novel approaches to treatment in the past 2 decades have been sparse. Since 2006, of 126 approvals granted by the US Food and Drug Administration for hematology/oncology medications, none have been for the treatment of bladder cancer, and chemotherapeutic approaches remain rooted in cisplatin-based combinations first introduced 30 years ago. This limited progress has provided major incentive to analyze molecular alterations in bladder cancer in detail in an effort to identify novel treatment approaches.
Bladder cancer genetics and molecular biology have historically provided important general insights into cancer biology, beginning with the discovery of HRAS as the first oncogene in a bladder cancer cell line. Since that seminal discovery, multiple genes commonly subject to mutation in bladder cancer have been identified, including TP53 , RB1 , TSC1 , FGFR3 , and PIK3CA . Furthermore, comparative genomic hybridization and related techniques were used extensively in bladder cancer, leading to identification of multiple amplified and deleted genes, including PPARG , E2F3 , EGFR , CCND1 , and MDM2 , which are amplified, and CDKN2A and RB1 , which are commonly deleted. These and other molecular alterations involved in bladder cancer have been summarized in previous reviews.
Recently, next-generation sequencing has enabled large-scale analyses, mainly focused on muscle-invasive bladder cancer, greatly expanding our understanding of this malignancy. The initial next-generation sequencing studies were performed by the Beijing Genomics Institute, in studies that focused initially on mutation identification, and then included both mutation analysis and transcriptome studies. More recently, The Cancer Genome Atlas (TCGA) project, funded by the National Cancer Institute, has performed a comprehensive analysis of 131 muscle-invasive bladder cancers, including assessment of mutations, copy number changes, expression profiling by RNA-Seq, micro RNA (miRNA) analysis, CpG methylation analysis, proteomic analysis of about 150 proteins, and integrated analyses of these data sets.
This review summarizes the current understanding of molecular alterations in bladder cancer, and focuses on findings from the TCGA project and the Beijing group. We also discuss recent reports providing improved understanding of molecular subtypes of bladder cancer based on expression analyses.
Background
Bladder cancer is a leading cause of morbidity and mortality, with nearly 400,000 new cases and 150,000 deaths worldwide. However novel approaches to treatment in the past 2 decades have been sparse. Since 2006, of 126 approvals granted by the US Food and Drug Administration for hematology/oncology medications, none have been for the treatment of bladder cancer, and chemotherapeutic approaches remain rooted in cisplatin-based combinations first introduced 30 years ago. This limited progress has provided major incentive to analyze molecular alterations in bladder cancer in detail in an effort to identify novel treatment approaches.
Bladder cancer genetics and molecular biology have historically provided important general insights into cancer biology, beginning with the discovery of HRAS as the first oncogene in a bladder cancer cell line. Since that seminal discovery, multiple genes commonly subject to mutation in bladder cancer have been identified, including TP53 , RB1 , TSC1 , FGFR3 , and PIK3CA . Furthermore, comparative genomic hybridization and related techniques were used extensively in bladder cancer, leading to identification of multiple amplified and deleted genes, including PPARG , E2F3 , EGFR , CCND1 , and MDM2 , which are amplified, and CDKN2A and RB1 , which are commonly deleted. These and other molecular alterations involved in bladder cancer have been summarized in previous reviews.
Recently, next-generation sequencing has enabled large-scale analyses, mainly focused on muscle-invasive bladder cancer, greatly expanding our understanding of this malignancy. The initial next-generation sequencing studies were performed by the Beijing Genomics Institute, in studies that focused initially on mutation identification, and then included both mutation analysis and transcriptome studies. More recently, The Cancer Genome Atlas (TCGA) project, funded by the National Cancer Institute, has performed a comprehensive analysis of 131 muscle-invasive bladder cancers, including assessment of mutations, copy number changes, expression profiling by RNA-Seq, micro RNA (miRNA) analysis, CpG methylation analysis, proteomic analysis of about 150 proteins, and integrated analyses of these data sets.
This review summarizes the current understanding of molecular alterations in bladder cancer, and focuses on findings from the TCGA project and the Beijing group. We also discuss recent reports providing improved understanding of molecular subtypes of bladder cancer based on expression analyses.
Molecular alterations in bladder cancer
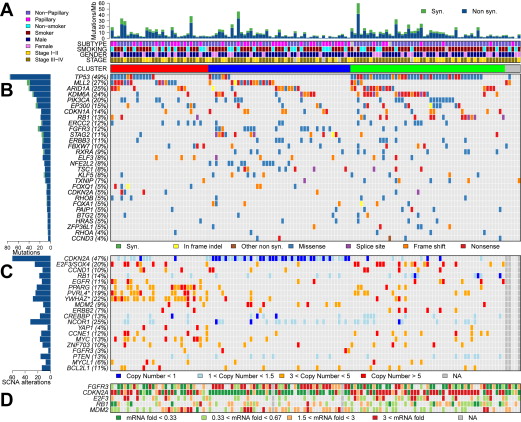
Fig. 1 illustrates the major findings of the TCGA study, showing mutation rates and frequencies, gene deletions and amplifications, and changes in expression for genes of interest.
Mutations in Bladder Cancer: General Findings
The TCGA study identified a relatively high rate of 7.68 mutations per Mb within coding regions, equivalent to 302 exonic mutations per cancer. This mutation rate is exceeded among cancers studied in the TCGA project (now >20) only by lung adenocarcinoma, lung squamous cell carcinoma, and melanoma. The mechanism or cause of this high mutation rate in bladder cancer is not known with certainty. Although smoking is associated with mutation rate and spectrum in lung cancer, this was not seen in bladder cancer, despite the well-known epidemiologic association between cigarette smoking and bladder cancer. On the other hand, 51% of mutations seen in bladder cancer were TCW -> TTW or TGW changes (“TCW mutations” [C > T and C > G mutations] at T-C-A/T [TCW] trinucleotides), a class of mutation likely mediated by one of the DNA cytosine deaminases, in the APOBEC gene family. In addition, APOBEC3B was expressed at high levels in all bladder cancers examined, suggesting a major role for APOBEC-mediated mutagenesis in the high mutation rate seen in bladder cancer.
The Beijing group identified a somewhat lower overall mutation rate, but by statistical analyses identified significant levels of mutation in 37 genes. This included many genes identified previously, as well as multiple chromatin remodeling genes, namely, KDM6A , ARID1A , CREBBP , EP300 , KMT2A , NCOR1 , CHD6 , and KMT2C . In the TCGA analysis, 32 genes were identified as sustaining mutations at a significant rate (see Fig. 1 B). There was considerable overlap between the genes identified by the Beijing group and those identified in the TCGA analysis, providing significant confirmation, despite the distinctly different populations being studied (Beijing, Chinese; TCGA, mostly American and of European origin). Genes sustaining significant levels of mutation can be organized into several classes.
Somatic copy number alterations in bladder cancer
Somatic copy number alterations (SCNAs) were also common in the TCGA data set, with an average of 204 segmental SCNAs and 22 genomic rearrangements per cancer analyzed. Statistical methods (GISTIC) were used to identify significant recurrent focal SCNAs, and 27 amplified and 30 deleted regions were identified (see Fig. 1 C). CDKN2A was deleted in nearly half of the samples, and other genes that were probable targets of deletion included KDM6A , RB1 , WWOX , PDE4D , FOXQ1 , FAM190A , LRP1B , and CREBBP . Many deletions extended over a large genomic region and a single gene target could not be identified. Many genes were focally amplified, including E2F3/SOX4 , CCND1 , EGFR , PPARG , MDM2 , ERBB2 , YAP1 , CCNE1 , MYC , ZNF703 , FGFR3 , MYCL1 , and BCL2L1 . Other chromosomal regions of amplification extended over a large genomic region, containing more than a single gene. This included a region on chromosome 1q22–23.2 containing PVRL4 , and a region on chromosome 8q22.3 containing YWHAZ . Multiple past reports made similar, although more limited, findings.
Cell-cycle gene aberrations in bladder cancer
Mutation or deletion of cell-cycle genes in cancer has been known for many years, and reported previously in bladder cancer for over 2 decades (for examples ). The recent in-depth genomic analyses performed have confirmed and extended our knowledge of cell-cycle gene events in bladder cancer. Reviewing these alterations in the order of frequency, TP53 (encodes p53) mutations were found in 49% and 24% of bladder cancers by the TCGA and Beijing groups. p53 is the “guardian of the genome,” and it responds to cellular stress by inducing cell-cycle arrest, apoptosis, senescence, and DNA repair. Its loss leads to bypass of these important effects, enhancing further genome damage, and continued proliferation. CDKN2A deletions are also very common in bladder cancer, similar to many other malignancies, with deletions seen in about 50% of samples in both the TCGA and Beijing studies. Mutations in CDKN2A were also seen in 5% of bladder cancers. CDKN2A encodes both p19 ARF and p16 INK4A proteins, which regulate the p53 and RB pathways, respectively. p16 INK4A is a cyclin-dependent kinase (cdk) inhibitor for cdk4 and cdk6, and its loss enhances cell-cycle progression.
RB1 was mutated in 10% and deleted in 15% of TCGA tumors, with similar findings by the Beijing group. The RB protein regulates the cell cycle by binding to the E2F transcription factor, and its loss also leads to enhanced cell-cycle progression despite DNA damage or other signals.
A novel finding in the TCGA analysis was inactivation of CDKN1A by mutation in 14% and deletion in 6%. Another group reported similar findings simultaneously. CDKN1A encodes another cyclin-dependent kinase inhibitor, p21, distinct from p16 INK4A , but its loss similarly pushes the cell toward continued division despite DNA damage or other signals, enhancing cell proliferation and accumulation of additional DNA damage. CDKN1A mutation has not been seen at appreciable frequency in any other cancer studied to date by the TCGA.
Three cell-cycle genes were amplified in bladder cancer in these studies: CCND1 (cyclin D1) in 10% to 21% of bladder cancers, CCNE1 (cyclin E1) in 12% to 13%, and MDM2 in 9% to 4% of samples, respectively, in the TCGA and Beijing analyses. Both cyclin D1 and cyclin E1 are necessary cofactors for different cdks and hence their overexpression enhances cell proliferation. MDM2 encodes an E3 ubiquitin–protein ligase, the enzyme that degrades p53 protein; hence, its overexpression lowers p53 levels, thereby inhibiting p53 function. MDM2 amplification was mutually exclusive of TP53 mutation for the most part in the TCGA analysis (see Fig. 1 ).
Overall, 1 or more cell-cycle genes were mutated, deleted, or activated by amplification in 93% of the TCGA tumor samples, most often the TP53 and/or CDKN2A genes (see Fig. 1 ).
Kinase signaling: receptor tyrosine kinases and the phosphatidylinositol-3-kinase-mammalian target of rapamycin pathway
Given the well-known successes of tyrosine kinase inhibitor therapy for activated kinases in both chronic myelogenous leukemia (driven by BCR-ABL) and lung adenocarcinoma (driven by activating mutations in EGFR ), there is major interest in the identification of similar activated tyrosine kinases in all types of cancer. Activating mutations in FGFR3 were first identified in bladder cancer 15 years ago ; they are more commonly seen in superficial than in muscle-invasive cancers. FGFR3 mutations were seen at 11% frequency in the muscle-invasive cancers studied by the TCGA and Beijing groups. In addition, 3% of cases had FGFR3 amplification, and another 3% had FGFR3-TACC3 gene fusions. These latter gene fusions have recently been reported by several groups, and shown to be activating. Three members of the ERBB family of receptor tyrosine kinases were also altered at significant frequency in bladder cancer. EGFR amplification was seen in 11%, ERBB2 was mutated in 5% and amplified in 7%, and ERBB3 was mutated in 11% and amplified in 2% of bladder cancers. These events in FGFR3 and ERBB family members are all potentially targetable using either tyrosine kinase inhibitors, or antibody-mediated therapies. This potential is underscored by a recent report of the benefit of both erlotinib (EGFR tyrosine kinase inhibitor) and cetuximab (monoclonal antibody against EGFR) in some subsets of muscle-invasive bladder cancers.
Mutations involving genes in the phosphatidylinositol-3-kinase (PI3K)–protein tyrosine phosphatase (PTEN)–AKT–TSC1–TSC2–mammalian target of rapamycin (mTOR) signaling pathway have long been recognized in bladder cancer. The recent large-scale studies provided further documentation of mutation of both positive and negative regulators in this signaling cascade. PIK3CA was mutated in 15% and amplified in 5%, PTEN was mutated in 3% and deleted in 13%, TSC1 was mutated in 8%, and TSC2 was mutated in 2% in the TCGA analyses, with similar results from the Beijing studies. All of these mutations lead to potential therapeutic targets, as highlighted by 2 recent studies showing that bladder cancer patients with TSC1 and MTOR mutations were exceptional responders to treatment with/including everolimus, an mTOR allosteric inhibitor.
Other genes and pathways
Two genes involved in the response to oxidative stress were found to be mutated in bladder cancer in the TCGA analysis. NFE2L2 encodes a transcription factor that is induced to mediate the cellular response to oxidative stress, and missense mutations in this gene were identified in 8% of the TCGA samples. Mutations in NFE2L2 were previously reported in cancer and shown to promote malignancy, although not previously known for bladder cancer. TXNIP encodes thioredoxin interacting protein and is also involved in mediating the response to oxidative stress. Mutations were seen in 7% of cancers in the TCGA study.
Mutations in genes involved in lipid metabolism were also identified in the TCGA analysis. RXRA , which encodes the retinoid X nuclear receptor alpha, was mutated in 9% of bladder cancers, with 7 of 12 mutations occurring at the same amino acid in the ligand-binding domain. All 7 of these cancers showed increased expression of genes involved in lipid metabolism, consistent downstream effects of RXR activation. PPARG , which encodes the peroxisome proliferator-activated receptor gamma (glitazone receptor), was amplified in 17% of bladder cancers.
STAG2 is another relatively novel gene found to be mutated in bladder cancer by several groups at frequencies from 10% to 20%. The STAG2 protein is a subunit of cohesin, a protein complex that regulates the separation of sister chromatids during cell division. Conflicting results were seen in terms of a potential association between STAG2 mutation and survival in bladder cancer.
ERCC2 was observed to be mutated in 12% and 7% of bladder cancers in the TCGA and Beijing analyses. ERCC2 is a nucleotide excision repair gene that causes xeroderma pigmentosum. It seems that these mutations may act in a dominant negative fashion, because 15 of 16 were deleterious missense mutations. A recent report described a positive association between ERCC2 mutation and response to cisplatin-based chemotherapy in bladder cancer, consistent with loss of ERCC2 function leading to an Achilles heel-like sensitivity to cisplatin.
Epigenetics: Chromatin Regulation
The role of epigenetic effects on regulation of gene expression, and their alterations in cancer in general have been studied for many years. However, the potential importance of these events in bladder cancer was advanced considerably by the recent in-depth genomic analyses. Mutations in chromatin regulatory genes in bladder cancer were first identified by the Beijing group, and then confirmed and extended by the TCGA analyses. It is known that there are 2 main categories of chromatin modification that influence gene expression. The first is methylation at the C position of CG nucleotide sequences, and other less common modifications to the nucleotide sequence itself. The second is histone modifications, which are generated by chromatin regulatory genes. Chromatin regulatory genes are often referred to as “writers,” “erasers,” and “readers,” based on their function in creating covalent modification of histones (methylation, acetylation), removing such modifications (demethylation, deacetylation), and in binding to such modifications to influence gene transcription. Genes involved in all 3 of these functions are commonly mutated in bladder cancer.
Mutations in KDM6A , an “eraser” demethylase acting on histone H3 at lysine 27, are seen in 20% to 25% of bladder cancers. MLL2 , encoding a histone 3 lysine 4 (H3K4) methyltransferase of the trithorax group, is a “writer,” and is mutated in 27% of bladder cancers. ARID1A , encoding a member of the SWI/SNF family, has helicase and ATPase activity, is a “reader,” and is mutated in 25% of bladder cancers. EP300 , encoding a histone acetylase, is a “writer,” which is also a transcription factor, and is mutated in 15% of bladder cancers. Many other chromatin regulatory genes are mutated in more than 10% of bladder cancer samples, including MLL3, MLL, CREBBP, CHD7 and SRCAP . In addition, CREBBP and NCOR1 were deleted in 13% and 25% of bladder cancers, respectively (see Fig. 1 ). Despite the likely importance of these mutations in the chromatin regulation of the genome in bladder cancer cells, there was no association between any of these gene mutations and expression profile or other features in the TCGA analysis.
Viral infection in bladder cancer: cytomegalovirus, BK polyomavirus, and human papillomavirus 16
Exploration of infectious etiologies has long been of interest in bladder cancer: A viral etiology for bladder cancer has been considered for some time, and there is a clear epidemiologic relationship to chronic Schistosoma infection. The TCGA analysis identified 7 tumors (6%) with viral DNA sequences and five tumors with viral RNA transcripts. The tumors with viral transcripts were: 3 with cytomegalovirus, 1 with BK polyomavirus, and 1 with human papillomavirus 16. Some of those with viral DNA sequences showed integration of viral DNA into the genome, with 1 instance of integration into BCL2L1 , an apoptosis regulatory gene, suggesting that insertional mutagenesis might contribute to bladder cancer development in these cases.
Molecular Subtypes of Bladder Cancer
There is considerable heterogeneity in bladder cancer, both in terms of natural history and response to chemotherapy. Many efforts have been made to define molecular subsets of bladder cancer based on mutation profile and/or expression features to attempt to provide prognostic information, as well as guidance in selection of chemotherapy. For example, Takata and colleagues analyzed gene expression profiles of biopsy materials from 27 invasive bladder cancers by microarray to identify 14 “predictive” genes whose expression levels were most correlated with response to methotrexate, vinblastine, doxorubicin, and cisplatin (M-VAC) chemotherapy. Ongoing studies are attempting to validate this expression profile prospectively. In the TCGA analysis, unsupervised clustering by non-negative matrix factorization of mutations and focal SCNAs identified 3 groups (see Fig. 1 A). Group A ( red ), labeled “focally amplified,” was highly enriched in focal SCNAs in several genes, as well as mutations in MLL2 . Group B ( blue ), labeled “papillary CDKN2A -deficient FGFR3 mutant,” was enriched in papillary histology, and the majority had loss of CDKN2A , and 1 or more alterations in FGFR3 . Group C ( green ), labeled “ TP53 /cell-cycle mutant,” had TP53 mutations in nearly all samples, and enrichment for RB1 mutations, and amplifications of E2F3 and CCNE1 (see Fig. 1 ).
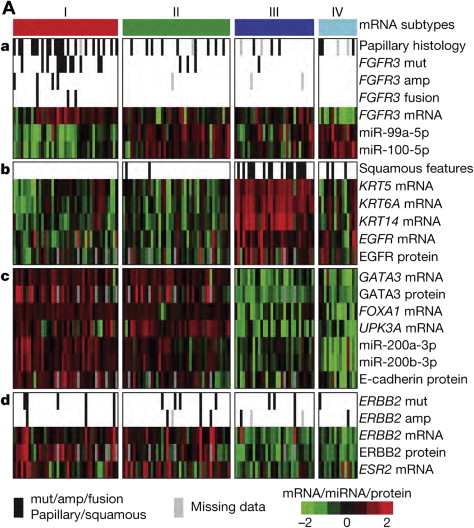
The TCGA analysis also used RNA-Seq based expression profiling to identify 4 distinct mRNA expression clusters ( Fig. 2 A). Cluster I (“papillarylike”) was enriched in cancers with papillary morphology, FGFR3 mutations, FGFR3 copy number gain, elevated FGFR3 expression, and FGFR3 – TACC3 gene fusions. Cluster II was similar to cluster I in several respects, but did not have an association with papillary morphology or FGFR3 events. Clusters I and II both expressed high HER2 ( ERBB2 ) levels and had an elevated estrogen receptor beta ( ESR2 ) signaling signature, suggesting a relationship to HER2-positive breast cancers. Clusters I and II also had features similar to those of luminal A breast cancer, with high expression of GATA3 and FOXA1 . I and II also expressed uroplakins (markers of urothelial differentiation), E-cadherin, and members of the miR-200 family of miRNAs (which target multiple regulators of epithelial–mesenchymal transition), suggesting that these cancers had urothelial differentiation to some extent. In contrast, cluster III (“basal/squamous-like”) was similar in some respects to both basal-like breast cancer, and squamous cell cancers of the head and neck and lung with expression of epithelial lineage genes, including several keratin genes. Some also showed some degree of variant squamous histology on pathology review.
Related posts:
 Bladder Cancer
Bladder Cancer
 New Trends in the Surgical Management of Invasive Bladder Cancer
New Trends in the Surgical Management of Invasive Bladder Cancer
 Second-Line Therapies in Metastatic Urothelial Carcinoma
Management of Low-risk and Intermediate-risk Non–Muscle-invasive Bladder Carcinoma
First-Line Treatment and Prognostic Factors of Metastatic Bladder Cancer for Platinum-Eligible Patients
First-line Treatment of Metastatic Disease
Second-Line Therapies in Metastatic Urothelial Carcinoma
Management of Low-risk and Intermediate-risk Non–Muscle-invasive Bladder Carcinoma
First-Line Treatment and Prognostic Factors of Metastatic Bladder Cancer for Platinum-Eligible Patients
First-line Treatment of Metastatic Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


