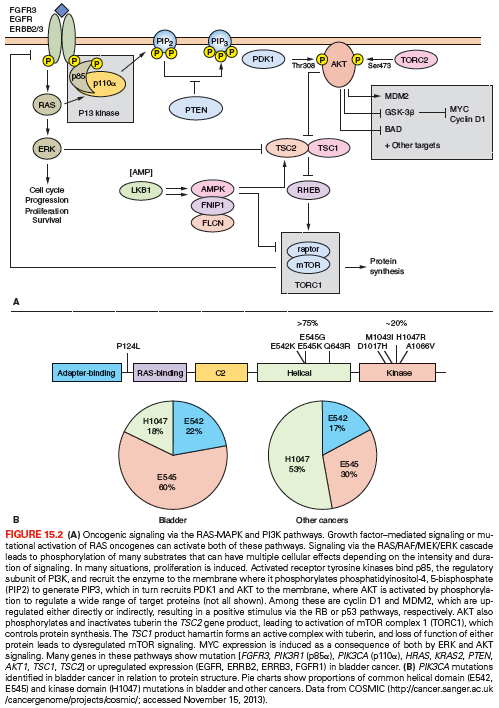
Activation of the RAS-MAPK pathway in Ta tumors may also be achieved by mutation of one of the RAS genes (most commonly HRAS or KRAS2), and this is mutually exclusive with FGFR3 mutation.15 More than 80% of noninvasive bladder tumors are predicted to have RAS-MAPK pathway activation via these mechanisms (Fig. 15.2A). Compatible with this, urothelial expression of an activated Ras transgene in mice leads to hyperplasia and papillary tumors,16 suggesting an important role for activation of the RAS-MAPK pathway in the generation of urothelial hyperplasia.
In NMI UC, FGFR3 mutation is associated with favorable outcome.17–19 High FGFR3 expression, normal staining pattern for CK20, and low proliferative index in papillary urothelial neoplasms of low malignant potential 20 is reported to identify tumors that do not recur.21
FGFR3 is considered a good therapeutic target in superficial UC, though early clinical application is most likely in muscle invasive rather than superficial UC (see the following). Several studies indicate that inhibition of mutant FGFR3 by knockdown or inhibition using small molecules or antibodies has a profound effect on UC cell phenotype, including inhibition of xenograft growth in vivo.22
Upregulated expression of the related receptor FGFR1 is also found in Ta tumors.23 No mutations have been detected, and there is infrequent gene amplification.24 Ectopic expression of FGFR1 in NHUC in the presence of FGF2 ligand, activates the RAS-MAPK pathway and PLCγ, and promotes cell survival.23 Currently, there is no information on the prognostic significance of these alterations.
PIK3CA
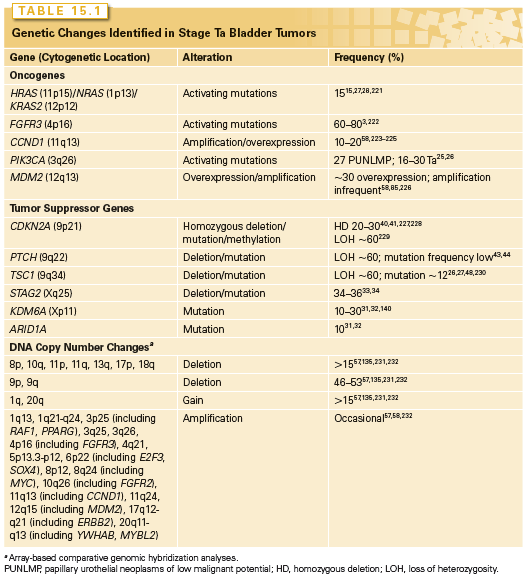
The phosphatidylinositol-3 kinase (PI3K) pathway plays a pivotal role in signaling from receptor tyrosine kinases (Fig. 15.2A). Activating mutations of the p110α catalytic subunit (PIK3CA) are found in UC, most commonly in low-grade, stage Ta tumors (~25%).25–28 The mutation spectrum (Fig. 15.2B) differs significantly from that found in other cancers. Mutations E542K and E545K in the helical domain are most common (22% and 60%, respectively) and the kinase domain mutation H1047R, which is the most common mutation in other cancers, is less frequent. The selective pressure for helical domain mutation in UC is not fully understood. E542K and E545K forms require interaction with RAS-GTP but not binding to p85, the regulatory subunit of PI3K, whereas H1047R depends on p85 binding and is active in the absence of RAS binding.29 This suggests potential cooperation between helical domain PIK3CA mutant proteins and events in UC that activate RAS. Compatible with this, PIK3CA and FGFR3 found together.25–28 Mutant PIK3CA confers a proliferative advantage at confluence and stimulates intraepithelial movement in NHUC, with higher activity of helical domain than kinase domain mutants in this cell context.30 Several other mechanisms of PI3K pathway activation have been identified in UC, though none of these are common in noninvasive tumors (Table 15.1).
STAG2
Inactivating mutations in the cohesin complex component STAG2 (Xq25) have been identified in UC.31–34 Mutations are most common in stage Ta tumors (20% to 36%) and are predominantly inactivating, suggesting a tumor suppressor function. The cohesin complex, which in human cells contains SMC1A, SMC3, RAD21, and either STAG2 or STAG1, mediates cohesion between sister chromatids following DNA replication to ensure correct chromosomal segregation. Mutations in STAG2 in glioblastoma have been reported to cause aneuploidy35 but this relationship is not apparent in UC, where most mutations have been found in low grade/stage, genomically stable tumors, and there is no association of mutation with chromosomal copy number changes.31,34
In addition to its well-documented functions during cell division, cohesin regulates gene expression through mechanisms involving DNA looping and interactions with transcriptional regulators such as CTCF, though evidence to date suggests that these roles are mainly related to STAG1-cohesin.36 Functional studies are now required to elucidate the consequences of STAG2 loss of function in UC.
Telomerase Reverse Transcriptase Gene Promoter
The most common genomic alterations identified in UC of all grades and stages are mutations in the promoter of the telomerase reverse transcriptase gene (TERT) in more than 80% of cases.37,38 Mutations are predominantly in two hotspot positions [-124 bp (G>A) and -146 bp (G>A) relative to the ATG translational start site], and this has facilitated the design of robust methods of detection. Examination of TERT expression in UC tissues has not revealed an effect of mutation on expression,37 so that the functional significance of these mutations remains to be determined. Nevertheless, the ease with which these mutations can be detected in urine sediments37,38 is likely to make a major contribution to the development of urine-based assays for detection of bladder tumors of all grades and stages.
Other Genomic Alterations in Noninvasive Urothelial Carcinoma
These tumors are often near diploid. The most common genomic alteration is loss of heterozygosity (LOH) or copy number loss of chromosome 9, often an entire homolog. More than 50% of UC of all of grades and stages show chromosome 9 LOH. A critical region on 9p21 and at least three regions on 9q (9q22, 9q32–q33, and 9q34) have been identified. Candidate genes within these regions are CDKN2A (p16/p14ARF) and CDKN2B (p15) at 9p21,39–42 PTCH (Gorlin syndrome gene) at 9q22,43,44 DBC1 at 9q32–q33,45–47 and TSC1 (tuberous sclerosis syndrome gene 1) at 9q3426,48,49 (Table 15.1).
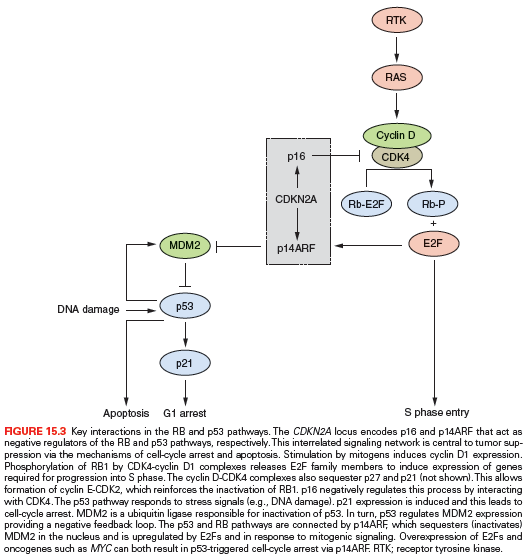
CDKN2A (9p21) encodes the two cell-cycle regulators, p16 and p14ARF. p16 is a negative regulator of the RB pathway and p14ARF, a negative regulator of the p53 pathway (Fig. 15.3). Inactivation of this locus in UC is commonly by homozygous deletion (HD). LOH of 9p, HD of CDKN2A, and loss of expression of p16 in NMI UC is predictive of reduced recurrence-free interval.50–52 Mouse knockout and in vitro experiments indicate that p16 and/or p14ARF may be haploinsufficient.53,54 In human UC, this may affect the biology of approximately 45% of cases that have LOH or reduced copy number of 9p21.
On 9q, three genes are implicated. PTCH, the Gorlin syndrome gene (9q22), shows infrequent mutation,44 but many tumors have reduced mRNA expression.43 DBC1 (9q33) shows HD in a few tumors55 and no mutations, but is commonly silenced by hypermethylation.45,56 TSC1 is the best-validated 9q tumor suppressor gene. TSC1 in complex with TSC2 negatively regulates the mTOR branch of the PI3K pathway (Fig. 15.2A).
Biallelic inactivation of TSC1 is found in 12% to 16% of UC with no relationship to grade or stage.26,27
Cyclin D1
CCND1 (11q13) is amplified in some superficial and invasive UC,57,58 and the protein is overexpressed in an even larger number.59,60 Overexpression in many cases may be the consequence of other alterations, such as activation of the MAPK or PI3K pathways (Fig. 15.2A). In Ta tumors, upregulated expression is associated with higher risk of progression to muscle invasion.60
KEY MOLECULAR ALTERATIONS IN INVASIVE UROTHELIAL CARCINOMA
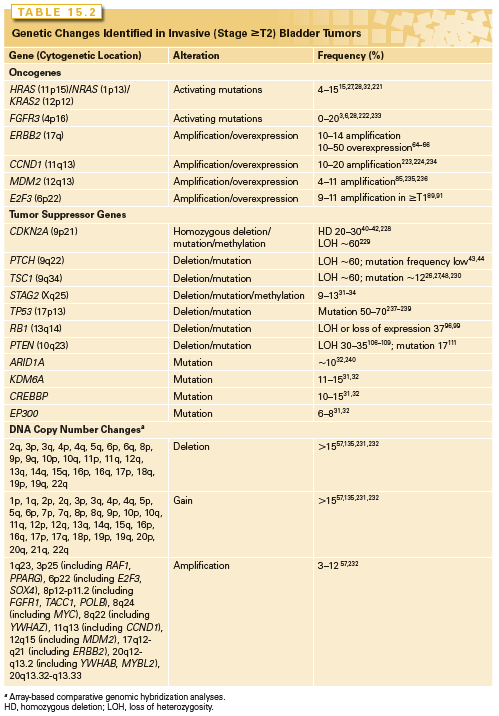
Many genomic alterations are found in muscle-invasive (MI) UC, including alterations to known genes and genomic alterations for which the target genes are currently unknown (Table 15.2).
Oncogenes
Overexpression of EGFR, ERBB2, and/or ERBB3 is associated with higher tumor grade and stage and with clinical outcome.61–63 ERBB2 (17q23) is amplified in 10% to 20% and overexpressed in 10% to 50% of MI UC.64–66 Amplification is more common in metastatic lesions than in the related primary tumor.67 As this receptor cannot bind ligand and relies on heterodimerization with ERBB3, it is likely that ERBB3 status and/or ligand expression may have significant influence.65,68,69 Up to 70% of MI tumors overexpress EGFR, and this is associated with poor prognosis.61,70,71 Both ERBB2 and EGFR represent potential therapeutic targets in advanced UC.72 These changes may activate the RAS-MAPK and/or PI3K pathways (Fig. 15.2A).
RAS gene mutation is not associated with either invasive or noninvasive disease (mutations in ~13% overall).15 Although mice expressing mutant H-ras in the urothelium develop superficial papillary tumors rather than MI tumors,73 in vitro experiments in human tumor cells indicate that HRAS can induce an invasive phenotype.74 Thus, RAS mutation may contribute to development of both major forms of UC.
PIK3CA and FGFR3 are mutated less frequently than in NMI UC. Approximately 15% of T2 tumors show FGFR3 mutation.3,6,75 However, protein expression is upregulated in 40% to 50% of non-mutant MI UC.6 Thus FGFR3 is considered a good therapeutic target in invasive and metastatic disease and preclinical studies have shown that gene knockdown76 or treatment with FGFR-selective small molecules and antibodies inhibits cell proliferation and tumorigenicity of UC cell lines with mutant or upregulated FGFR3.77–79 Potential predictive biomarkers include mutation, overexpression, or detection of an FGFR3 fusion protein. The presence of FGFR3 fusion proteins in UC cell lines10 is associated with good response to FGFR inhibitors.77 Epithelial phenotype may provide an additional biomarker as in vitro assays indicate that UC cells with epithelial phenotype have enhanced sensitivity to FGFR inhibition compared to those with mesenchymal phenotype.80 A recent RNAi screen in UC cell lines indicated that upregulated EGFR signaling provides a mechanism of escape from FGFR inhibition and can mediate de novo resistance. A reciprocal relationship was found when EGFR was inhibited and both in vitro and in a preclinical in vivo model, combined inhibition showed improved anti-tumor activity.81 Thus, assessment of both FGFR3 and EGFR status may be required to predict response and combined EGFR and FGFR3 inhibition may be essential. Several clinical trials of FGFR inhibitors are now planned or in progress in advanced UC.
FGFR1 is also overexpressed in many of these cancers.23 An increased ratio of the FGFR1-β : FGFR1-α splice variants is found in MI tumors. The β isoform, lacking the first immunoglobulin-like domain, has increased sensitivity to FGF1.82 FGF2 stimulation of ectopically expressed FGFR1-β in some UC-derived cell lines induces an epithelial-mesenchymal transition (EMT), a major feature of which is PLCγ-mediated upregulation of COX-2.83 This is in contrast to the effect of FGFR1 signaling in NHUC, where increased proliferation and reduced apoptosis but no EMT is induced,23 suggesting that FGFR1 plays different roles in NMI and MI UC. Compatible with this, UC cell lines with highest FGFR1 expression show a mesenchymal (EMT) phenotype (low E-cadherin expression) and upregulated FGF2 expression, and those with epithelial phenotype show higher FGFR3 and E-cadherin and lower FGFR1.84 In an animal model of UC metastasis using an FGFR1-dependent cell line, an FGFR inhibitor reduced the development of circulating tumor cells and metastasis but not primary tumor growth.84 Currently, there is no information on the prognostic significance of FGFR1 upregulation, and FGFR1 has not yet been examined independently of FGFR3 as a potential therapeutic target.
Several other oncogenes are implicated in MI UC. Four to six percent have amplification of MDM2 (12q14).57,85,86 MDM2 regulates p53 levels, and overexpression provides an alternative mechanism to inactivate p53 function (Fig. 15.3). There is no consensus on the relationship of upregulated MDM2 to tumor grade, stage, or prognosis. MYC is upregulated in many bladder tumors, although the mechanism for this is unclear.87 Although amplifications of 8q are found in invasive UC, MYC is not the major target. However, additional copies of the whole of 8q are common and may lead to overexpression.24,57 MYC is also upregulated in response to other molecular events (e.g., MAPK pathway stimulation). An amplicon on 6p in 14% of MI UC and cell lines contains E2F3, and functional studies indicate that E2F3 can drive urothelial cell proliferation.88–92 E2F transcription factors interact with and are regulated by RB1 (Fig. 15.3) and in accord with this, tumors with E2F3 amplification have RB1 or p16 inactivated.89
Tumor Suppressor Genes
As in other aggressive cancers, the tumor suppressor genes TP53, RB1, CDKN2A, and PTEN are implicated in MI UC. The pathways controlled by TP53 and RB1 regulate cell-cycle progression and responses to stress (Fig. 15.3). TP53 mutation is common in invasive UC and detection of mutation or TP53 protein accumulation is associated with poor prognosis. Although immunohistochemical detection of TP53 protein with increased half-life identifies many mutant TP53 proteins and is commonly used as a surrogate marker for mutation, some TP53 mutations (~20%) yield unstable or truncated proteins that cannot be detected in this way. Thus, TP53 protein accumulation is not a useful prognostic marker. Two meta-analyses indicate only a small association between TP53 positivity and poor prognosis.93,94 However, examination of both protein expression and mutation of TP53 provides useful prognostic information.95
The RB pathway regulates cell-cycle progression from G1 to S phase (Fig. 15.3). HD, LOH of 13q14, and loss of RB1 protein expression are common in MI UC.96–99 Loss of p16 expression is inversely related to positive RB1 expression,100 and high-level p16 expression results from negative feedback in tumors with loss of RB1.101 Thus, both loss of expression and high-level expression of p16 are associated with RB pathway deregulation, and these are adverse prognostic biomarkers found in >50% of MI tumors.102 Interestingly, in MI UC with FGFR3 mutation, a high frequency of CDKN2A HD has been reported, which may identify a progression pathway for NMI FGFR3-mutant tumors to muscle invasion via loss of CDKN2A.103 As indicated previously, amplification and overexpression of E2F3, which is normally repressed by RB1, is associated with RB1 or p16 loss in MI tumors.89 p16 and p14ARF proteins link the RB and p53 pathways (Fig. 15.3), and due to multiple regulatory feedback mechanisms, inactivation of both pathways together is predicted to have greater impact than inactivation of either pathway alone. This is borne out by the achievement of greater predictive power in studies using concurrent analyses of multiple changes that deregulate the G1 checkpoint.102,104,105
Altered PTEN expression is the most frequent mechanism for deregulation of the PI3K pathway in MI UC. PTEN (phosphatase and tensin homolog deleted on chromosome 10) (10q23) is a lipid and protein phosphatase whose major lipid substrate is phosphatidylinositol (3,4,5)-triphosphate (PIP3) generated by PI3K (Fig. 15.2A). PTEN LOH is found in 24% to 58% of invasive UC,106–108 and copy number analysis has identified a relationship of PTEN loss to metastatic disease.57 Mutation of the retained copy is infrequent but HD has been detected in tumor cell lines.26,109–111 Overall, 46% of UC lines (largely derived from MI tumors) had PTEN alterations.26 Reduced expression is common in tumors26,112 and is associated with TP53 alteration. Many tumors (41%) with altered TP53 show downregulation of PTEN, and this combined alteration is associated with poor outcome.113 As PTEN is haploinsufficient in mouse models,114 loss of one allele in UC may lead to altered phenotype. In mice, conditional deletion of Pten in the urothelium led to early urothelial hyperplasia112,115 and late development of tumors resembling human papillary superficial tumors. A study that induced stochastic deletion of p53 and/or Pten showed that deletion of either gene alone did not lead to tumor formation, but dual deletion led to early development of aggressive UC, with frequent metastases.113 PTEN loss may affect proliferation, apoptosis, and migration. Reexpression in PTEN-null UC cells has revealed effects on cell chemotaxis, anchorage-independent growth, and tumor growth in vivo.116–118 UC cell invasion can be inhibited by the protein phosphatase activity of PTEN alone.116 Thus, loss of lipid and protein phosphatase activities of PTEN may contribute in different ways to urothelial tumorigenesis.
The TSC1 product hamartin acts in the PI3K pathway downstream of PTEN (Fig. 15.2A), providing an alternative mechanism of pathway activation in 13% of invasive UCs that have mutational inactivation.26,27 Whereas good response to mTOR inhibitors has been reported in some patients and UC cell lines with mutant TSC1, mutation alone is not a sufficient predictive biomarker.119,120
The Rho GDP dissociation inhibitor RhoGDI2 has been implicated as a tumor suppressor in UC. Expression is reduced in an isogenic cell line model of metastasis and low expression is associated with reduced survival in patients with UC.121 Potential downstream effectors of RhoGDI2 are endothelin and versican, which are upregulated upon loss of RhoGDI2 expression. In an experimental model of metastasis, endothelin 1 was required for lung colonization by UC cells and pharmacologic inhibition of the endothelin axis reduced colonization.122 RhoGDI2-regulated versican levels are implicated in enhancing macrophage recruitment and chemokine CCL1 levels.123 As both of these proteins enhance the inflammatory response, the major role of RhoGDI2 may be to inhibit this. RhoGDI2 is phosphorylated by SRC, and this enhances its membrane localization and metastasis-suppression activity.124 Compatible with this, and unlike the situation in many tumors, SRC levels in UC are highest in NMI tumors where RhoGDI2 function is intact.125,126 SRC contributes to suppression of metastasis via regulation of p190Rho GAP, which in turn downregulates the activity of RHOA, RHOC, and ROCK.127
The WNT signaling pathway is also implicated in UC. Mutations in APC have been reported,27,128 mainly in MI tumors. The frequency of mutations in CTNNB1 is low, but altered beta-catenin expression (reduced expression or increased nuclear localization) is frequent in MI UC,128–131 though this is not associated with APC mutation.128 Other alterations in the WNT pathway include epigenetic silencing of the antagonists of WNT signaling, secreted frizzled receptor proteins,132 and the Wnt inhibitory factor 1 (WIF1).133
Other Genomic Alterations in Invasive Urothelial Carcinoma
Invasive UC is often genomically unstable, with significant genetic divergence of related tumors in the same patient over time. Array-based comparative genomic hybridization and single nucleotide polymorphism array analyses have identified many copy number alterations57,134–137 (Table 15.2). These include numerous losses and gains in DNA copy number and many regions of high-level amplification that may contain novel oncogenes. To date, the target genes within many of these regions have not been identified. Many regions of copy number alteration show significant association with high tumor grade, stage, and/or outcome. These include amplicons at 1q21–24, 3p25, 6p22, 8p11, 8q22, 11q12, 12q15, and 17q12. Plausible candidate oncogenes within these are PPARG (3p25), E2F3 and SOX4 (6p22), FGFR1, TACC1 and POLB (8p11), YWHAZ (8q22), CCND1 (11q12), MDM2 (12q15), and ERBB2 (17q12).57,135,138,139 On 1q21–24, at least three regions have been defined, two of which contain plausible candidates (BCL9 and CHD1L in one, and MCL1, SETDB1, and HIF1B in a second).139 Regions of HD include 1p34, 2q36, 9p21, 11p11, 18p11, and 19q13.57 On 9p22, CDKN2A is one target and two further regions of HD have been identified.57 Candidates within the other regions are less obvious.
It is notable that some stage T1 tumors show similar profiles to MI tumors (T2 or greater), suggesting that these tumors with the ability to break through the basement membrane may be aggressive lesions. However, other T1 tumors show remarkable similarity in their copy number profiles to Ta tumors, suggesting that distinct biologic subgroups exist.57
INFORMATION FROM EXOME SEQUENCING
At the time of writing, three studies have reported sequencing of whole UC exomes.31,32,140 In total, these have sequenced 12 Ta, 41 T1, and 72 T2 tumors. In two studies, selected genes were assessed in larger sample sets to examine prevalence.31,140 Whereas the overall numbers of tumors studied remains relatively small, several important conclusions can be drawn. The first is that UC contains a relatively high frequency of somatic single nucleotide mutations, with estimates ranging from 50 to 170 per sample on average but with wide variability between samples. Many of the mutations (≥ 30%) were nonsynonymous, a large proportion of which are predicted to be damaging (~60% estimated in one study31). To date, significant differences in mutation rates between non-aggressive and aggressive tumors are not apparent, though it is notable that very few stage Ta tumors have been studied so far. The most common base change reported is C:G>T:A transition followed by C:G>G:A transversion.
The most significant finding is that in addition to genes already implicated in UC (TP53, FGFR3, HRAS, KRAS, PIK3CA, RB1, TSC1), a large number of genes involved in chromatin modification show mutation. In the largest study, 58% of tumors had mutation in chromatin remodeling genes,32 indicating that altered regulation of chromatin is a major oncogenic driver in UC. Mutated genes include histone demethylases (KDM6A, KDM5A, KDM5B, UTY), chromatin remodeling genes (ARID1A, ARID4A), histone lysine methyltransferases (MLL, MLL2, MLL3, MLL5), histone acetyltransferases (CREBBP, EP300, EP400), and SWI/SNF complex-related genes (SMARCA4, SMARCA1). Many of the mutations are predicted to impair function, suggesting tumor suppressor roles. Mutations were also found in DNA repair genes including ATM, FANCA, and ERCC2. Overall, in MI tumors the most frequently mutated genes were KDM6A (30%), TP53 (24%), ARID1A (15%), CREBBP (15%), EP300 (13%), HRAS (13%), RB1 (13%), PIK3CA (12%), STAG2 (11%), and FGFR3 (11%).32 In all three studies, relatively large numbers of genes were mutated at low frequency, suggesting that there is considerable biologic heterogeneity.
EPIGENETIC ALTERATIONS
Epigenetic alterations, mainly those involving DNA methylation, have been widely reported. Most studies have analyzed single genes or small series of genes.141–144 Genome-wide analyses have identified novel candidates, some with clinicopathologic associations.145–149 In general, hypermethylation in CpG islands is more common in MI tumors and hypomethylation in regions distinct from CpG islands in NMI tumors.147,148 Integrated analysis of methylation and gene expression indicates that CpG island hypermethylation is associated with loss of expression and CpG methylation within genes or at transcription factor binding sites with increased expression.149 Hypermethylation-associated disease progression biomarkers in NMI tumors include TBX2, TBX3, TBX4, GATA2, and ZIC2.145,147 These genes, which encode transcription factors involved in lineage decisions during development, are implicated in EMT, stem cell phenotype, and differentiation. In the high-risk group of T1 grade 3 tumors treated with BCG, combined methylation of MSH6 and THBS1 is associated with disease progression.150 Hypermethylation of several miRNAs is associated with tumor grade and stage. Some of these have prognostic value (e.g., the miR-200 family and miR-205) and are frequently silenced in MI tumors, and this is correlated with disease progression in T1 tumors.151
There are many reports of improved detection of UC by analysis of methylation biomarkers in urine. Examples include a panel of five biomarkers (MYO3A, CA10, NKX6-2, DBC1, and SOX11 or PENK),152 two panels each of four biomarkers (ZNF154, POU4F2, HOXA9, and EOMES),147 (APC, 2 TERT CpGs, and EDNRB),153 one of two markers (TWIST1 and NID2),154 and one of three markers (OTX1, ONECUT, and OSR1), used in combination with FGFR3 mutation.155
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


