Fig. 2.1
Simplified cellular organization
The aim of this chapter is to highlight some of the molecular abnormalities involved in carcinogenesis, and potential challenges to finding perfect ‘anti-cancer drugs.
Carcinogenesis
The molecular causes (causative agents) of cancer can broadly be classified into the following three categories:
1.
Genetic (e.g., activation of oncogenes, inactivation of tumor suppressor genes, germline mutation)
2.
Epigenetic (e.g., abnormal chromatin remodeling/histone modifications, mitochondrial damage, abnormal gene silencing)
3.
Systemic/paracrine (abnormal growth factors/receptors, hormonal abnormalities, inflammatory cytokines/chemokines)
Additionally, several environmental causative agents, such as ultraviolet (UV) light, numerous chemicals, and radioactive agents may interact with the different molecular causative agents leading to a series of events involved in the initiation and progression of cancer. Some of these events include the following [9]:
1.
Loss of normal signals for cell proliferation leading to uncontrolled proliferation
2.
Loss of normal signals for cell differentiation
3.
Sustained cell division
4.
Avoidance/evasion of apoptosis
5.
Ability to invade basement membrane and other tissues
6.
Angiogenesis
In addition to these events, cellular microenvironment also plays a role in carcinogenesis. A single event is usually not sufficient for carcinogenesis, and in fact the majority of the events listed above are necessary for carcinogenesis following insults from causative agent(s) [9]. A detailed discussion of the causative agents and events in carcinogenesis is beyond the scope of this chapter. Our focus will be a concise discussion of the common causative agents and events. While the discussion is divided into sections, it is important to understand that the events leading to carcinogenesis are complex, interrelated, and cannot be compartmentalized. We will begin by discussing what happens to the cell as a whole and what eventually happens at the molecular level (Fig. 2.1).
Cellular Injury
Eukaryotic cells are constantly being exposed to both endogenous and exogenous injurious agents that affect cell structure and function. Endogenous sources of injury include spontaneous reactions intrinsic to the chemical nature of DNA in an aqueous solution which may lead to deamination (that is, removal of an amine group) [3], reactive oxygen and nitrogen species, lipid peroxidation products, endogenous alkylating agents, and other molecules formed from normal oxidative body metabolism [3, 10, 11]. Exogenous agents include the environmental causative agents mentioned previously, which to some extent are avoidable. The effects of all of these may lead to DNA strand breaks or spontaneous base loss as high as 104–105 per cell per day [3]. The cumulative effects of all these insults may range from minor to major cellular damage. The cellular damage may be repaired or may initiate series of events leading to cell death or uncontrolled proliferation. Thus, cells need extensive repair mechanisms to counteract the effects of constant cell injury inducing agents. Some of the repair mechanisms including mismatch repair (MMR), nucleotide excision repair (NER), double-strand break repair (DSBR), and base excision repair (BER) will be discussed later. However, the body’s repair mechanisms may be overwhelmed by extensive or prolonged injury leading to the development of cancer. For example, iron overload in the liver, chronic inflammation in the gastrointestinal system, chronic obstructive pulmonary disease (COPD), and asbestosis [12–16] can lead to hepatocellular carcinoma, gastrointestinal carcinoma, lung carcinoma, and mesothelioma respectively.
Chronic inflammation or irritation may cause an abnormality in the epithelial barriers and apparent compartmentation in the gastrointestinal tract [12, 13, 17] leading to an aberrant expression or interaction of factors and receptors normally required for growth and repair. For example, in the gastrointestinal tract, the epidermal growth factor (EGF) is reportedly secreted across the apical membrane whereas the EGF receptor (EGFR) is located in the baso-lateral cell surfaces to prevent continuous or persistent interaction. EGF normally only has access to the EGFR in the case of an injury [17–19] which temporarily enables EGF binding to EGFR to facilitate the repair process. EGF’s access and binding to EGFR ceases when the cause of injury is eliminated and repair is complete. Prolonged injury or constant irritation in this case may lead to abnormal proliferation due to continuous or persistent EGF-EGFR binding.
Cell Membrane Channels
Channels in the cell membrane, such as sodium-potassium channels, are necessary for normal cellular functions and homeostasis. Abnormalities of some of these channels may be associated with cancer. For example, aquaporin, present in several tissues, is a class of membrane water channel which facilitates passive transport of water across cell membrane osmotic gradient. Abnormalities of this have been reportedly involved in tumor angiogenesis, invasion, metastasis, and altered signaling pathways [20–27]. While there is ongoing research, there is currently no significant clinical application in care of cancer patients.
Cell Membrane Receptors
The central dogma of factor/ligand and receptor interaction indicating that receptors are mostly inactive in the absence of a factor/ligand (Fig. 2.2) is useful for understanding pathways to carcinogenesis [28] and will be the focus of the following discussion.
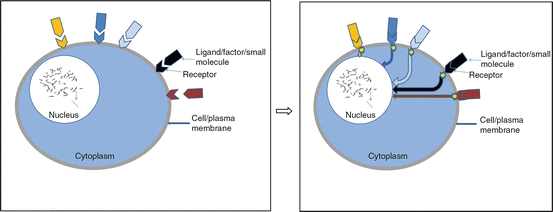
Fig. 2.2
Central dogma of ligand-receptor interaction for activation of pathways
Binding of a ligand/factor to its receptor leads to activation of the signaling pathways mostly through kinases. Factors/ligands in the body bind to specialized targets on the cellular membrane (receptors) to initiate a cascade of intracellular processes (signaling pathways) resulting in repair, cell proliferation/growth, or cell death [29] (see Fig. 2.3). These ligands/factors are mostly growth factors and include epidermal growth factor (EGF), vascular endothelial growth factor A (VEGFA), platelet-derived growth factor alpha polypeptide (PDGFA), and insulin-like growth factors (IGF) among others. The respective membrane receptors with intrinsic tyrosine kinase (TK) activity [29–31] include kinase insert domain receptor (KDR)/VEGFA receptor (KDR aka VEGFR) and EGF receptors (EGFR) especially EGFR (aka ERBB1). These receptors appear to be the dominant receptors in solid tumors. The most frequently occurring carcinomas have overactive kinases secondary to cell membrane protein/receptor activation [29, 32–34] predominantly through EPH receptor B2 (EPHB2 aka ERK) and v-akt murine thymoma viral oncogene homolog 1 (AKT1 aka AKT). Following binding of receptor tyrosine kinase to appropriate ligand/factor, there is reorganization of the receptors and autophosphorylation of the intracellular portion of the molecules. Although, ligand-receptor interaction is the central dogma, some receptors such as v-erb-b2 avian erythroblastic leukemia viral oncogene homolog 2 (ERBB2 aka HER2) have no known ligand and can be activated by dimerization with another EGFR [35, 36].
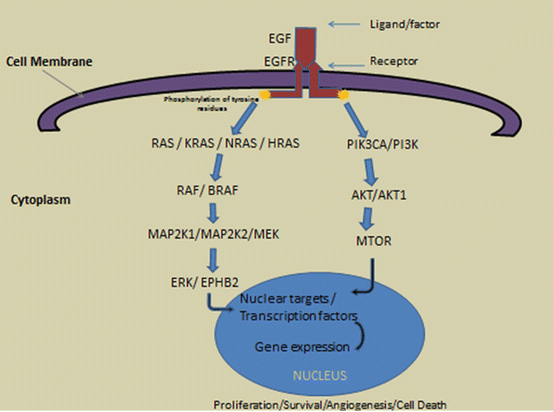
Fig. 2.3
Simplified central dogma of factor or ligand-receptor interaction and the activate cascade/pathway using EGF-EGFR as a prototype. Gene products in the pathway: KRAS kirsten rat sarcoma viral oncogene homolog, NRAS neuroblastoma RAS viral (v-ras) oncogene homolog, RAS rat sarcoma viral oncogene, HRAS Harvey rat sarcoma viral oncogene homolog, BRAF B-Raf proto-oncogene, serine/threonine kinase, RAF Raf proto-oncogene, MAP2K1 mitogen-activated protein kinase kinase 1, MAP2K2 mitogen-activated protein kinase kinase 2, EPHB2 EPH receptor B2, PIK3CA phosphatidylinositol-4,5-bisphosphate 3-kinase, catalytic subunit alpha, PI3K phosphoinositide-3-kinase, AKT1 v-akt murine thymoma viral oncogene homolog 1, MTOR mechanistic target of rapamycin (serine/threonine kinase)
For the signaling pathways to be effective, its activities need to be transported to the nucleus (Fig. 2.3) by different cellular trafficking mechanisms to induce transcription [37–42]. The nuclear targets of the signaling pathways include chromatin, cyclin D1 (CCND1), jun proto-oncogene (JUN), v-myc avian myelocytomatosis viral oncogene homolog (MYC), FBJ murine osteosarcoma viral oncogene homolog (FOS), v-myb avian myeloblastosis viral oncogene homolog (MYB), prostaglandin-endoperoxide synthase 2 (PTGS2 aka COX2), and signal transducer and activator of transcription 3 (STAT3) among many others [42–44].
Although the illustrations of factor-receptor pathway (Figs. 2.2 and 2.3) above are simplified, the reality is much more complex. First, while normal cells use the same pathway for physiologic activities, abnormalities in this process may lead to abnormal growth (cancer) or cell death. Second, cancer cells generally express more than one receptor whose activation may or may not require binding to ligands. Furthermore, activation of these receptors can all induce downstream signaling pathways through downstream kinases with some overlap or interaction (crosstalk) between different signaling pathways [32, 45]. As such, developing an ideal therapeutic target is challenging and development of therapeutic resistance is not uncommon. Nevertheless, ongoing research is focused on overcoming or minimizing resistance to targeted cancer therapies, and some targeted therapies that are currently in clinical use (Table 2.1) or in clinical trials (Table 2.2) were developed based on the knowledge of these signaling pathways [46, 47]. The mechanisms of action of some of the targeted therapies are highlighted in Fig. 2.4.
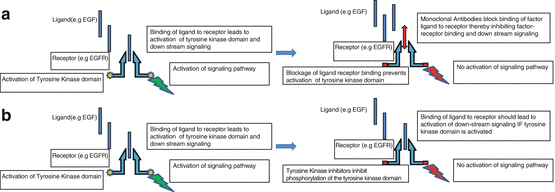
Table 2.1
Some common cancer target therapies
Small molecules/factors/ligands | Cell membrane receptors involved | Common cancers | Targeted treatment in clinical use/clinical trials |
|---|---|---|---|
No ligand | ERBB2 (aka HER2) | Breast cancer, gastric, esophageal adenocarcinoma | ERBB2/HER2 antibody Trastuzumab/Herceptin |
Anti-EGFR & ERBB2 Lapatinib | |||
EGF | EGFR | Lung cancer, colon cancer, prostate, cervical cancer | Anti-EGFR Cetuximab |
Anti-EGFR Erlotinib | |||
Anti-EGFR/ TK inhibitor gefitinib | |||
Anti-EGFR antibody SYM004 | |||
VEGFA (aka VEGF) | KDR (aka VEGFR) | Colorectal cancer | Anti-VEGFR2 Ramucirumab |
Anti-VEGFR2/PDGFR Sorafenib | |||
Anti-VEGF Bevacizumab | |||
Anti-VEGF Aflibercept (Zaltrap) | |||
MET/VEGFR2 inhibitor GSK1363089 | |||
KITLG (aka mast cell growth factor) | KIT (aka mast cell growth factor receptor) | Gastrointestinal stromal tumors, ovarian tumors, acute myeloid leukemia (AML), systemic mastocytosis, and melanoma | Tyrosine kinase inhibitor Imatinib |
Tyrosine kinase inhibitor Nilotinib | |||
PDGFA | PDGFRA | Gastrointestinal stromal tumors | Anti-VEGFR2/PDGFR Sorafenib |
Anti-PDGFR Crenolanib | |||
Anti-PDGFR Fovista (E10030) | |||
Multitargeted RTK inhibitor MGCD516 |
Table 2.2
Some targeted therapies in clinical trials
Gene mutation | Tumor and % of occurrence | Targeted treatment in clinical use or in clinical trials |
|---|---|---|
KRAS (activated by point mutation, gene amplification, and other means) | Pancreas (90 %); colorectal (45 %); papillary thyroid (60 %); follicular thyroid (55 %); seminoma (45 %); NSCLC (35 %) | MEK inhibitor—MSC1936369B, |
MAPK inhibitor/MEK inhibitor AZD6244 | ||
BRAF (activated by point mutation) ~7 % of all cancers | Melanoma (27–70 %); papillary thyroid (36–53 %); ovarian serous papillary (30 %); colorectal (5–22 %) | BRAF inhibitor Vemurafenib |
BRAF inhibitor GSK2118436/Dabrafenib | ||
MEK inhibitor GSK1120212/Trametinib | ||
PIK3CA (activated by point mutation, gene amplification) >30 % of all cancers | Breast (8–40 %); endometrial (26–36 %); hepatocellular (36 %); head and neck squamous cell (33 %); colorectal (19–32 %); prostate (29 %); GBM (5–27 %); gastric (25 %) | AKT inhibitor AZD5363 |
PI3K inhibitor BYL719 | ||
PI3K inhibitor BKM120/Buparlisib | ||
PI3K inhibitor GDC-0032 | ||
PIK3 inhibitor GDC-0941 | ||
PTEN (inactivated by deletion, methylation, protein stability) | Endometrial (38 %); CNS (20 %); skin (17 %); prostate (14 %); colon (9 %). | PI3K-beta inhibitor GSK2636771 |
PI3K-beta inhibitor AZD8186 | ||
AKT1 (AKT1 is infrequently mutated in human cancer but AKT2 gene can undergo mutation) | AKT1: thyroid (5 %), breast (3 %), endometrial (3 %), ovary (1 %), urinary tract (1 %), prostate (1 %), large intestine (1 %), hematopoietic and lymphoid tissue (1 %) | AKT inhibitor (AZD5363) |
AKT inhibitor GSK2141795 | ||
AKT inhibitor MK2206 | ||
AKT inhibitor GDC-0068 | ||
AKT2: Head and neck squamous cell carcinomas (30 % amplified), pancreatic (20 % amplified), ovarian (12 % amplified), breast (3 % amplified) |
Fig. 2.4
Mechanisms of action of (a) monoclonal antibodies (e.g., cetuximab) and (b) tyrosine kinase inhibitors (e.g., erlotinib)
Changes Occurring at the Genetic Level (DNA Level)
The ultimate target of cellular injury or damage is the DNA machinery in the nucleus. Defective DNA, if unrepaired, may lead to defective transcripts, defective gene products, and abnormal cellular function, the consequence of which may be uncontrolled proliferation and tumor formation. There is an important difference between DNA damage and mutation. DNA damage is an undesired but repairable alteration in base, sugar, or phosphate leading to alteration of the properties of the DNA [2]. Mutations on the other hand are fixed errors in the coding information [2, 3]. Damage, if unrepaired or incorrectly repaired, may lead to incorporation of an incorrect base leading to permanent damage and subsequently mutation. The cell cycle plays a significant role in preventing cells with significant damage from dividing precluding passing the damage to daughter cells.
The cell cycle is an intrinsic part of normal bodily functions with tight regulatory mechanisms, abnormalities of which may be catastrophic for cells. While the G1, S, G2, and M phase of the cell cycle is well known (Fig. 2.5), the complexity of cell cycle and regulatory mechanisms may be less appreciated. The cell cycle progresses from G1 phase through M phase. There are multiple damage response mechanisms (DRMs) to address DNA damage, ensuring that under normal circumstances, only normal cells complete M phase, while damaged cells are inhibited [45, 48]. To ensure the effectiveness of the DRMs, there are complex surveillance mechanisms, called checkpoints [48], such as G1/S, intra S, G2/S, and G2/M, which though distinct, share many proteins. For example a single-strand DNA damage may induce over 700 proteins, more than 900 phosphorylation and several ATP molecules [2, 3, 50, 51], all in an effort to maintain genomic integrity. The DRM surveillance mechanisms include sensor proteins which detect DNA damage (e.g., proliferating cell nuclear antigen [PCNA] and replicating factor C [RFC]-like protein complex); transducer protein (sends/transduces the signals to effector protein); the effector protein which via a series of event causes cell cycle arrest, apoptosis, DNA repair among others [48–51].
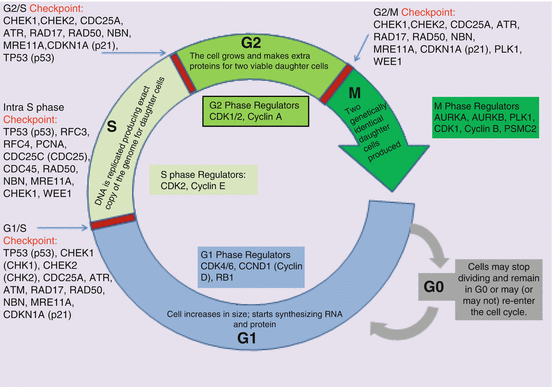
Fig. 2.5
Cell cycle, checkpoints, and regulations. CDC25A cell division cycle 25 homolog A, CDC45 cell division cycle 45, CDK cyclin-dependent kinase, CDKN1A cyclin-dependent kinase inhibitor 1A (p21, Cip1), CCND1 cyclin D1 (aka cyclin D/BCL), CHK1 checkpoint 1 kinase, CHK2 checkpoint 2 kinase, TP53 tumor protein p53 (aka p53), ATM ataxia telangiectasia mutated, PCNA proliferating cell nuclear antigen, RAD17 RAD17 homolog (previously RAD1), RFC3 replication factor C subunit 3, RFC4 replication factor C subunit 4, ATR ATR serine/threonine, NBS nibrin (previously known as NBS1), RAD50 RAD 50 homolog, MRE11A meiotic recombination 11 homolog A, PLK1 polo-like kinase 1, RB1 retinoblastoma 1, PSMC2 proteasome 26S subunit, ATPase 2, AURKA Aurora kinase A (Aurora A), AURKB Aurora kinase B (Aurora B), WEE1 WEE1 G2 checkpoint kinase
When there is DNA damage, checkpoint activation temporarily halts the cell cycle and triggers DNA repair mechanisms. If the DNA damage is irreversible, tumor protein 53 (TP53 aka p53) activates the apoptotic machinery. In this sense, TP53 behaves as guardian of the genome and prohibits defective cell to undergo replication. Of note, TP53 is a potent tumor suppressor (see section on Tumor suppressor). Defects in these checkpoint mechanisms or DRMs may allow cells with the defective DNA complete the cell cycle which may result initially in nonlethal mutations in daughter cells. Accumulations of these defects and mutations over time may lead to uncontrolled growth and tumorigenesis [52–54]. It is therefore not surprising that drugs targeting the cell cycle/DRMs (Table 2.3) are currently the subject of intense research and clinical trials [55–61].
Table 2.3
Some examples of cell cycle targeted therapy in solid tumors and the section of the cell cycle targeted
Name of drug | Targets | Tumor | Current clinical use | Clinical trials |
|---|---|---|---|---|
Taxanes (e.g., paclitaxel [Taxol] or docetaxel [Taxotere]) | Microtubules (Taxanes stop microtubules from assembling and disassembling) | Breast cancer | Breast cancer; gynecologic malignancies | Phase 2 |
Bortezomib | Proteasome enzyme complex inhibitor | Advanced solid tumors | Currently used for multiple myeloma and mantle cell lymphoma | Phase 1 (combination with Clofarabine) |
(LY2606368) | CHK1/2 inhibitor (CHK1 is a key kinase in intra S and G2/M checkpoint; CHK1 inhibitors sensitize TP53 mutant tumor cells to chemotherapeutic agents, radiation, or PARP inhibitors by abrogating the checkpoints and driving the cells into mitosis with unrepaired damage) | BRCA1/2 mutation associated breast or ovarian cancer, non-high risk triple negative breast cancer, and high grade serous ovarian cancer at low genetic risk | Phase 2 | |
Talazoparib | PARP inhibitor (PARP [poly ADP-ribose polymerase] is needed for DNA base excision repair. PARP inhibitors prevent PARP-mediated DNA repair of single-strand DNA breaks via the base excision repair pathway leading to DNA “collapse” and apoptosis) | Advanced and/or metastatic breast cancer patients with BRCA mutation (EMBRACA study) | Phase 3 | |
(BMN 673) | ||||
Olaparib | ||||
Ganetespib | Hsp90 inhibitors (bind to and inhibit Hsp90, causing proteasomal degradation of oncogenic proteins and the inhibition of cell proliferation) | Phase 1/2 | ||
Combination with the mTOR inhibitor sirolimus for patients with unresectable or metastatic malignant peripheral nerve sheath tumors | ||||
Indenoisoquinoline LMP400 | Topoisomerase inhibitors (Top1 inhibitor) form stable DNA-Top1 cleavage-complexes at unique DNA cleavage sites inducing replication- and transcription-mediated DNA damage and delay DNA repair, leading to apoptosis | Relapsed solid tumors and lymphomas | Phase 1 | |
MK-1775 | Anti-WEE1 (WEE1 encodes nuclear tyrosine kinase which catalyzes inhibitory phosphorylation of CDC2/cyclin B kinase leading to G2 cell cycle arrest in response to DNA damage via inactivation CDK1/CDC2-bound cyclin B; inhibition of WEE1 enhances antitumor effects of DNA damaging agents, specifically in TP53 negative tumors) | Advanced solid tumors | Phase 1 | |
GSK461364A | PLK1 inhibitor (PLK controls various steps in mitosis including spindle assembly, kinetochore-microtubule interaction, and cytokinesis; activation of APC/C [61]; inhibition disrupts mitosis/spindle assembly leading to cell death) | Advanced solid tumor and non-Hodgkin lymphoma | Phase 1 and phase 1/2 | |
NMS-1286937 | ||||
Alisertib | Aurora kinase A inhibitor (Aurora A is required for centrosome maturation and bipolar spindle assembly; inhibition disrupts mitosis) | Rhabdoid tumor | Phase 2 | |
GSK1070916A | Aurora kinase B/C inhibitor (Aurora kinase B is part of the chromosomal passenger complex, regulating chromosomal segregation and cytokinesis, when inhibited cells exit mitosis without proper chromosomal alignment resulting in polyploidy and eventual cell death) | Advanced solid tumor | Phase 1 |
Oncogenes
Proto-oncogenes are normally involved in processes governing cell proliferation and survival [62]. However, a proto-oncogene may be transformed into an oncogene by mutations, fusion/translocation, or amplifications, often with a gain-of-function [63]. The activation or transformation of oncogenes may lead to increased expression of a normal gene product, expression of more stable mutant protein, protein with altered functionality, or altered recruitment/subcellular localization of normal gene product [63]. The products of oncogenes include transcription factors, chromatin remodeler, growth factors, growth factor receptors, signal transducer, and apoptotic regulators (Table 2.4) [63]. The roles of oncogenes in carcinogenesis have been the subject of intense research, and some potential candidate drugs targeting oncogenes are under investigation [62, 64–67].
Table 2.4
Oncogenes, functions, and associated solid tumors
Proto-oncogene/oncogene (symbols) | Previously named/aka | Product/function | Method of activation | Solid tumor associated |
|---|---|---|---|---|
JUN | v-jun | Transcription factors | Deregulation | Sarcoma |
MYC | c-MYC | Transcription factors | Amplification | SCLC, breast cancer, esophageal cancer, cervical cancer, head and neck cancer |
v-MYC | ||||
MYCN | n-MYC | Transcription factors | Amplification | Neuroblastoma; lung carcinoma |
FOS | v-fos, c-fos, AP-1 | Transcription factors | Deregulation | |
EGF | Growth factor | |||
FGF4 | HST, HBGF | Growth factor | Constitutive production | Gastric cancer |
PDGFB | v-sis | Growth factor | Constitutive production | Glioma, fibrosarcoma |
EGFR | ERBB1 | Growth factor receptors | Amplification; point mutation | NSCLC; squamous cell carcinoma |
ERBB2 | HER2; NEU | Growth factor receptor | Amplification; point mutation | Breast; neuroblastoma, NSCLC |
RET | CDHF12, CDHR16, PTC, RET51 | Growth factor receptor | DNA rearrangement; point mutation, ligand-independent constitutive activation; fusion | Thyroid carcinoma—MEN2A, MEN2B |
MET | HGFR | Growth factor receptor | Rearrangement/ligand-independent constitutive activation | Hereditary papillary renal cell carcinoma; hepatocellular carcinoma |
HRAS | Membrane-associated G proteins | Point mutation | Colorectal, lung, pancreas | |
KRAS | KRAS1 | Membrane-associated G proteins | Point mutation | Colorectal, lung, melanoma, thyroid |
NRAS | N-ras | Membrane-associated G proteins | Point mutation | Colorectal, other carcinoma, melanoma |
BRAF | Membrane-associated G proteins | Point mutation | Colorectal, thyroid, ovary, melanoma | |
SRC | ASV, c-src | Signal transducer/cytoplasmic tyrosine kinase | Constitutive activation | Colorectal carcinoma |
YES1 | v-yes1 | Signal transducer/cytoplasmic tyrosine kinase | Constitutive activation | Sarcoma |
FGR | c-fgr, p55c-fgr | Signal transducer/cytoplasmic tyrosine kinase | Constitutive activation | Sarcoma |
BCL2 | Bcl-2, PPP1R50 | Inhibitor of apoptosis | Deregulated activity, gene amplifications | B-cell lymphoma |
While oncogenes have the capability to transform a cell, not all oncogenic activation leads to tumorigenesis as all oncogenes are not created equal [62]. Some oncogenes need cooperation/abnormalities of other genes to initiate cancer. For example: MYC (v-myc avian myelocytomatosis viral oncogene homolog) may need abnormalities of TP53 tumor suppressor gene; BCL2 (B-cell CLL/lymphoma 2) needs mutations in other oncogenes like MYC for cellular transformation (in lymphoma). Other oncogenes, on the other hand, activate several signal transduction pathways (e.g., Breakpoint Cluster Region-Abelson murine leukemia proto-oncogene 1, non-receptor tyrosine kinase BCR-ABL1 in chronic myeloid leukemia) and are less reliant on other genes or secondary events [62].
Tumor Suppressor Genes
Tumor suppressors are genes that protect the integrity of the genome [65, 68, 69] and balance the driving force of cell proliferation by the oncogenes. Genes responsible for keeping other genes healthy by preventing mutation or involved in DNA repair, (like tumor suppressor genes or some DNA repair genes) are termed caretaker genes. Familiar tumor suppressor genes include retinoblastoma 1 (RB1), tumor protein p53 (TP53 aka p53), and adenomatous polyposis coli (APC). Gatekeeper genes help control cell proliferation by its regulation of the cell cycle. Classic tumor suppressors like TP53 and RB1 are termed “gatekeepers” of the cell because they play a critical role in the cell cycle (see Fig. 2.5) in determining whether a cell progresses through the cell cycle or undergoes apoptosis [65]. Briefly, the activity of RB1 depends on its phosphorylation status (phosphorylated by CDK4/6 and dephosphorylated by phosphatase 1 alpha). Hypophosphorylated or unphosphorylated RB1 negatively regulates the activity of E2F1 (E2F transcription factor 1) leading to cell cycle inhibition [63]. TP53 under normal conditions has a short half-life of less than 30 min, but in response to damage it activates CDKN1A (p21) to halt cell cycle at G1/S checkpoint [63]. Some of the epigenetic modifiers that will be discussed later like DNA methyltransferases, DNA hydroxylase, histone acetyltransferases, histone methyltransferases, and chromatic remodeling proteins may affect the expression of tumor suppressor genes [63]. As shown in Fig. 2.5, when DNA is damaged, checkpoint activation temporarily halts the cell cycle and triggers DNA repair mechanisms. If the DNA damage is irreversible, cellular apoptotic mechanisms are triggered resulting in cell death. In this sense, tumor suppressor genes act as a guardian of the genome and prevent defective cell from replicating. Abnormalities of the tumor suppressor genes remove the “protective” role, which may lead to carcinogenesis.
The majority of sporadic cancers exhibit loss of function of TP53 activity due to deletion or mutations. TP53 germline mutations are identified in a familial cancer syndrome, referred to as Li-Fraumeni syndrome, in which the patients can have a spectrum of tumors, most common of which are bone and soft tissue sarcomas, adrenal cortical carcinomas, and brain tumors [70]. TP53 is inactivated by two important regulators, MDM2 proto-oncogene, E3 ubiquitin protein ligase (MDM2) and MDM4, p53 regulator (MDM4 also known as MDMX). Several studies have highlighted the importance of this TP53–MDM2–MDM4 loop in the initiation and development of wild-type TP53-containing tumors. Certain MDM2 inhibitors that interfere with the TP53–MDM2 interaction are currently in Phase I Clinical trials [71, 72].
Apoptosis and Carcinogenesis (Programmed Self-Death)
Apoptosis, otherwise known as programmed cell death, is a tightly regulated critical cellular process for eliminating damaged cells [73, 74]. It relies on a family of cysteine-dependent aspartic acid proteases (caspases), secreted as an inactive protein or zymogen, but becomes activated on dimerization or cleavage (Fig. 2.6).
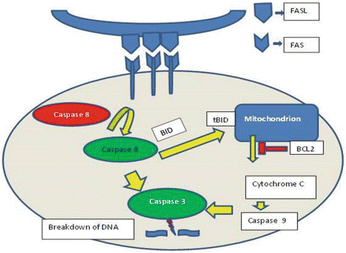
Fig. 2.6
Schematic diagram showing mechanisms of apoptosis. (BID BH3 interacting domain death agonist, FASLG (aka FASL) Fas ligand (TNF superfamily, member 6), FAS Fas cell surface death receptor, BCL2 B-cell CLL/lymphoma 2)
There are two main apoptotic pathways that are cysteinyl, aspartate-specific protease dependent (caspase-dependent) [74–76] pathways to apoptosis:
1.
Intrinsic or mitochondria apoptotic pathway: initiated by cellular stress, DNA damage causing alterations in the potential and permeability of the mitochondrial outer membrane. It involves a balance between the pro- and antiapoptotic B cell CLL/lymphoma 2 (BCL2) gene family, such as BCL2-like 1 (BCL2L1 aka BCLX), BCL2-associated X protein (BAX), and BCL2-antagonist/killer 1 (BAK1 aka BAK).
2.
Extrinsic or death receptor-mediated apoptotic pathway: initiated by activation of “death” the tumor necrosis factor (TNF) receptor family by respective ligands resulting in the recruitment of death-inducing signaling complex (DISC). BIRC2 (baculoviral IAP repeat containing 2; aka c-IAP1) and BIRC3 (baculoviral IAP repeat containing 3; aka c-IAP2) regulate the extrinsic pathway through ubiquitin ligase activity.
A comprehensive discussion of the mechanisms of apoptosis and regulation of apoptosis is beyond the scope of this chapter. Nonetheless, it is important to appreciate that abnormalities of this process have been associated with many human diseases including cancer. If apoptosis is inhibited, there may be accumulation of abnormal or damaged cells which may lead to cancer. Avoidance of apoptosis is one of the hallmarks of cancer and inhibitors of apoptosis (IAP) proteins prevent apoptosis, thereby promoting uncontrolled growth, and also play an important role in resistance to cancer therapy. Hence, the process of apoptosis, the effects of inhibitors of apoptosis (IAP), and drugs targeting IAP [74–80] have been the subjects of intense research. Thus targeted therapy to block IAP proteins is an attractive cancer treatment strategy [81]. In fact, Birinapant, a second generation antagonist of IAP proteins, that mimics “second mitochondrial-derived activator of caspases” (SMAC) activity is currently undergoing clinical trials for the treatment of solid tumors and hematological malignancies [82].
DNA Damage Repair Genes
A variety of DNA repair mechanisms exist to cope with the myriads of DNA or chromosomal damages [83, 84]. These include mismatch repair (MMR), nucleotide excision repair (NER), recombination repair, double-strand break repair (DSBR), and base excision repair (BER). If the repair system is overwhelmed by massive cellular/DNA damage or if the repair system is not properly maintained, DNA damage accumulates and can lead to cell death or cancer. Cancer cells appear to have defective repair mechanisms or have developed the ability to bypass the repair mechanisms [85]. Several human disorders with increased risks of cancers such as Xeroderma pigmentosum and Ataxia telangiectasia are caused by underlying defects in DNA repair genes [86, 87]. Although an activating mutation of an oncogene or an inactivating mutation or loss of a tumor suppressor gene may be a major event in the initiation of carcinogenesis, defective repair mechanisms enhance genomic instability and facilitate increased proliferation of precancerous cells. Figure 2.7 highlights the interrelationships of oncogenes, tumor suppressor, regulator of apoptosis, and DNA repair genes.
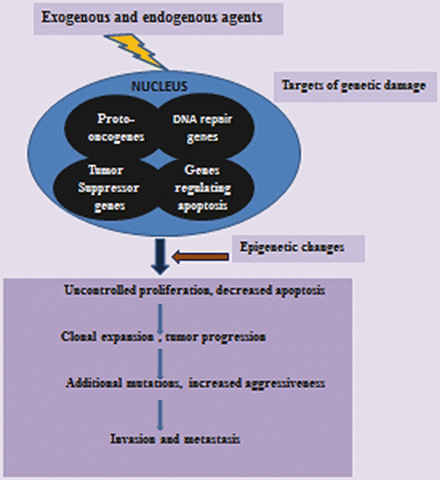
Fig. 2.7
Schematic diagram showing molecular basis of carcinogenesis
DNA Damage Repair System
MMR
Mismatch repair or MMR genes repair base–base and insertion/deletion (I/D) mismatches that escape the proofreading function of replicative polymerases. In addition, these genes have other important cellular functions including double-strand base repair, apoptosis, and anti-recombination. Common MMR genes include mutS homolog 2 (MSH2) and mutL homolog 1 (MLH1), mutS homolog 6 (MSH6), PMS2 (post-meiotic segregation increased 2 (S. cerevisiae)), and mutL homolog 3 (MLH3). MSH2 protein forms heterodimer with MSH6 or MSH3 to identify mismatches. MLH1 dimerizes with PMS2 to coordinate the binding of other proteins involved in mismatch repair [87–89]. The excision of the DNA strand containing the mispaired base is performed by exonuclease I and the re-synthesis is then performed by a polymerase [88, 90] as highlighted in Fig. 2.8.
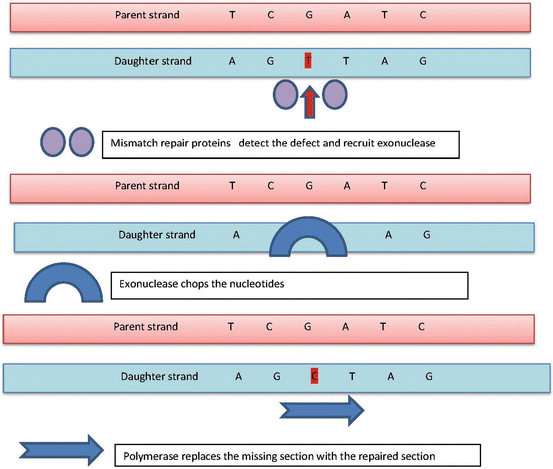




Fig. 2.8
Mismatch repair proteins first identify the site of mismatch (e.g., instead of C in the daughter strand, there is T) and then recruit the exonuclease enzyme, responsible for chopping or excising the mismatched nucleotide (sometimes chopping the nearby matched nucleotides), the polymerase then replaces the excised nucleotides with correct or matched one(s)
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
