In spite of a rapidly expanding understanding of head and neck tumor biology and optimization of radiation, chemotherapy, and surgical treatment modalities, head and neck squamous cell carcinoma (HNSCC) remains a major cause of cancer-related morbidity and mortality. Although our biologic understanding of these tumors had largely been limited to pathways driving proliferation, survival, and differentiation, the identification of HPV as a major driver of HNSCC and genomic sequencing analyses has dramatically influenced our understanding of tumor biology and approach to therapy. Here, we summarize molecular aspects of HNSCC biology and identify promising areas for potential diagnostic and therapeutic agents.
Key points
- •
Head and neck squamous cell carcinoma (HNSCC) is driven by numerous mutations, with human papilloma virus negative (HPV–) cancers caused by more traditional risk factors (tobacco use/alcohol) tending to harbor more mutations, greater intratumor heterogeneity, and extensive copy number variation.
- •
Recent genomic insights suggest that targeted therapy of HNSCC will remain a significant challenge. Most mutations identified based on sequencing analyses are loss-of-function mutations in known and putative tumor suppressor genes that may require novel approaches, such as synthetic lethality.
- •
Oncogenic drivers are few and far between and often are present at low mutant allele frequencies, suggesting they may be poor choices for targeted therapy.
- •
One exception for targeted therapy may be activating Ras or PI3K mutations that occur at high frequency in HPV+ cancers, offering a potential avenue for therapy that may facilitate deintensification of chemoradiation therapy.
- •
Identification of genes implicated in tumor-immune interactions as well as loss of function mutations suggest that immunotherapy and modulation of immune surveillance may be a valuable therapeutic approach, supporting ongoing immunotherapy clinical trials.
Introduction
Despite advances in our understanding of tumor biology, including its evolutionary refinements, as well as radiation, chemotherapy, and surgical treatments, head and neck squamous cell carcinoma (HNSCC) remains the sixth leading cause of cancer-related morbidity and mortality, with 550,000 new cases diagnosed each year. These tumors arise from mucosal epithelium in the oral cavity, oropharynx, larynx, and hypopharynx, which together represent 75% of diagnosed cancers.
HNSCC tumors can be broadly divided into those that are human papilloma virus (HPV)-negative (HPV–) and associated with alcohol and tobacco consumption, and those that are HPV-positive (HPV+) and due to HPV infection primarily with serotype 16. Although HPV– cancers arise via field cancerization and clonal progression in the setting of repetitive carcinogen application, HPV+ tumors harbor few mutations and are driven by a fundamentally distinct pathophysiologic mechanisms that rely on E6 and E7 viral proteins to inactivate or bypass cellular tumor suppressive responses. Although recent vaccines against HPV (Gardasil, Cervarix) will influence the prevalence of HPV+ HNSCC in the decades to follow, for now, the incidence of HPV+ HNSCC continues to rise. Current estimates suggest that 45% to 90% of oropharyngeal squamous cell carcinomas (OPSCCs) are HPV+ with 90% associated with HPV serotype 16. The division of HNSCC into two fundamentally distinct tumor cohorts with widely disparate survival rates based on HPV status represents one of the most significant developments of the past decade in head and neck cancer research and treatment.
Treatment for HNSCC is most often chosen based on the primary tumor subsite, TNM staging, and predicted functional outcomes following different treatment modalities. In general, early-stage (I or II) HNSCC is treated with local therapy, taking advantage of the ability of surgical removal or radiation to offer a curative modality. Advanced disease (stage III or IV) requires multimodality treatment with surgery, radiation, and/or chemotherapy. Although the influence of treatment-related medical complications on mortality has declined, and some improvements in head and neck survival have been documented, these are largely related to the increasing incidence of HPV+ cancers rather than substantive gain in the clinical management of HNSCC. Treatment failure in HNSCC relates to resistance of tumor cells to primary or adjuvant chemoradiation therapy, as well as residual undetectable microscopic disease that remains after surgical resection.
Recent whole-exome sequencing of HNSCC offers several lessons into how these tumors will need to be treated to improve on traditional therapeutic modalities. First, the sequencing of such a large number of tumors from numerous institutions demonstrates the successful endeavor of a multi-institutional collaborative effort to molecularly characterize the biology of head and neck tumors. Second, these analyses have validated that p53 inactivating mutations remain the predominant genetic defect identified, substantiating previous studies and emphasizing the observation that most tumors harbor loss-of-function mutations. Third, sequencing data separates HPV+ and HPV– tumors into distinct groups with completely different mutational profiles. Fourth, we have learned that HNSCC will be challenging to treat: there is no singular target for these tumors. Intratumor heterogeneity will also remain a challenge as we attempt to advance our therapeutic approaches.
In this review, we briefly discuss the molecular pathways driving HNSCC as identified using traditional genetics and biochemistry, but focus primarily on the new and interesting scientific advances in the field. In particular, we emphasize insights from recent whole-exome sequencing analyses of HNSCC, discuss lessons learned from analyses of intratumor heterogeneity, and explore the implications of recent studies on future therapeutic approaches.
Introduction
Despite advances in our understanding of tumor biology, including its evolutionary refinements, as well as radiation, chemotherapy, and surgical treatments, head and neck squamous cell carcinoma (HNSCC) remains the sixth leading cause of cancer-related morbidity and mortality, with 550,000 new cases diagnosed each year. These tumors arise from mucosal epithelium in the oral cavity, oropharynx, larynx, and hypopharynx, which together represent 75% of diagnosed cancers.
HNSCC tumors can be broadly divided into those that are human papilloma virus (HPV)-negative (HPV–) and associated with alcohol and tobacco consumption, and those that are HPV-positive (HPV+) and due to HPV infection primarily with serotype 16. Although HPV– cancers arise via field cancerization and clonal progression in the setting of repetitive carcinogen application, HPV+ tumors harbor few mutations and are driven by a fundamentally distinct pathophysiologic mechanisms that rely on E6 and E7 viral proteins to inactivate or bypass cellular tumor suppressive responses. Although recent vaccines against HPV (Gardasil, Cervarix) will influence the prevalence of HPV+ HNSCC in the decades to follow, for now, the incidence of HPV+ HNSCC continues to rise. Current estimates suggest that 45% to 90% of oropharyngeal squamous cell carcinomas (OPSCCs) are HPV+ with 90% associated with HPV serotype 16. The division of HNSCC into two fundamentally distinct tumor cohorts with widely disparate survival rates based on HPV status represents one of the most significant developments of the past decade in head and neck cancer research and treatment.
Treatment for HNSCC is most often chosen based on the primary tumor subsite, TNM staging, and predicted functional outcomes following different treatment modalities. In general, early-stage (I or II) HNSCC is treated with local therapy, taking advantage of the ability of surgical removal or radiation to offer a curative modality. Advanced disease (stage III or IV) requires multimodality treatment with surgery, radiation, and/or chemotherapy. Although the influence of treatment-related medical complications on mortality has declined, and some improvements in head and neck survival have been documented, these are largely related to the increasing incidence of HPV+ cancers rather than substantive gain in the clinical management of HNSCC. Treatment failure in HNSCC relates to resistance of tumor cells to primary or adjuvant chemoradiation therapy, as well as residual undetectable microscopic disease that remains after surgical resection.
Recent whole-exome sequencing of HNSCC offers several lessons into how these tumors will need to be treated to improve on traditional therapeutic modalities. First, the sequencing of such a large number of tumors from numerous institutions demonstrates the successful endeavor of a multi-institutional collaborative effort to molecularly characterize the biology of head and neck tumors. Second, these analyses have validated that p53 inactivating mutations remain the predominant genetic defect identified, substantiating previous studies and emphasizing the observation that most tumors harbor loss-of-function mutations. Third, sequencing data separates HPV+ and HPV– tumors into distinct groups with completely different mutational profiles. Fourth, we have learned that HNSCC will be challenging to treat: there is no singular target for these tumors. Intratumor heterogeneity will also remain a challenge as we attempt to advance our therapeutic approaches.
In this review, we briefly discuss the molecular pathways driving HNSCC as identified using traditional genetics and biochemistry, but focus primarily on the new and interesting scientific advances in the field. In particular, we emphasize insights from recent whole-exome sequencing analyses of HNSCC, discuss lessons learned from analyses of intratumor heterogeneity, and explore the implications of recent studies on future therapeutic approaches.
Tumor suppressors frequently drive head and neck squamous cell carcinoma but are difficult to target
p53 is a ubiquitous tumor suppressor that is critically altered in a number of human cancers, with up to two-thirds of HNSCCs harboring mutations in exons 5 to 8. Mutations in p53 dysregulate the cell cycle and monitoring of genomic integrity, thereby leading to aberrant proliferation, disrupted apoptosis, and defective DNA repair, whereas the HPV viral oncogene E6 targets p53 for degradation ( Fig. 1 ). Clinically, alterations in p53 function are associated with resistance to radiation and cisplatin-based chemotherapeutics, emphasizing the importance of this master regulator in HNSCC pathogenesis.
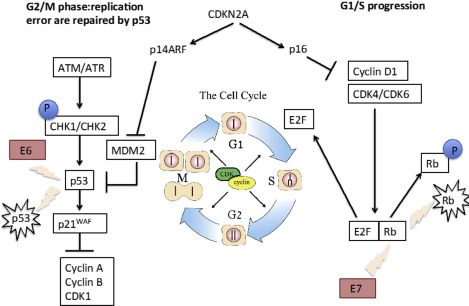
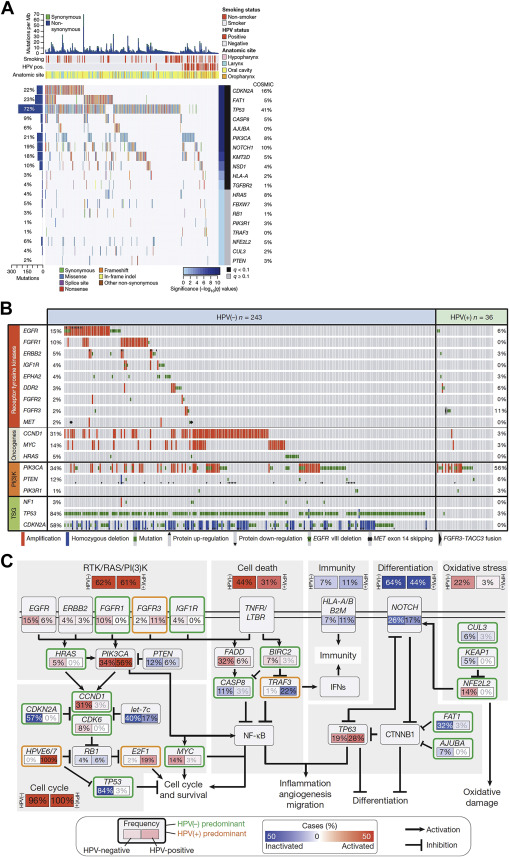
Recent whole exome sequencing analyses have validated these observations in cell lines and in vitro models, confirming that p53 mutations are common in HNSCC with loss-of-function mutations predominating. Stransky and colleagues analyzed 74 tumor-normal pairs with their analysis, suggesting 63% contained mutations or deletions in p53. Analyses from The Cancer Genome Atlas (TCGA) of 279 HNSCCs identified mutations in p53 in 84% of HPV– tumors, with only 3% (1 of 36) of HPV+ tumors containing a p53 mutation ( Fig. 2 ). Similarly, inactivating mutations in the cell-cycle regulator CDKN2A were found in 58% of HPV– tumors. Thus, a major conclusion of these whole-exome sequencing analyses has been validating the near-universal loss of function of p53 and CDKN2A inactivation in smoking/alcohol-related HNSCC. The challenge with p53and CDKN2A loss-of-function mutations is reactivation and/or replacing these critical cell-cycle regulators. Adenoviral gene therapy, chemical activators of mutated genes, and antagonists of endogenous p53 inhibitors are all possibilities, but preclinical and clinical trials hold variable promise, and these strategies suffer the inherent limitations of targeting tumor suppressor genes, including efficient delivery, tumor cell target specificity, and public resistance to gene therapy.
Alteration of differentiation pathways through the loss of transforming growth factor β receptor (TGFβR)/SMAD signaling may also promote the transformation of aerodigestive mucosa to invasive SCC by critically altering tumor suppressor pathways (see Fig. 2 ; Fig. 3 ). Loss of function mutations in TGFβR2 as well as in SMAD2 and SMAD4 have been identified. Interestingly, data from cutaneous SCC suggest that TGFβ may play a dual role in oncogenesis, initially acting as a tumor suppressor to prevent the transformation to invasive SCC, but subsequently promoting the epithelial-mesenchymal transition and supporting metastasis. Animal data from mice confirm this complex signaling dichotomy: conditional deletion of SMAD4 triggers genomic instability through activation of TGFβ1 and other SMADs, whereas deletion of TGFβR2 acts cooperatively with KRAS (V-Ki-ras2 Kirsten rat sarcoma viral oncogene homolog) to promote metastases. In whole-exome sequencing, comparison of mutations by subset analyses of anatomic site revealed unique mutations in TGFβR2 in oral cavity tumors, consistent with previously described functions in animal models. Given that TGFβ inhibitors are readily available and already being used in clinical trials for non–small cell lung cancer, colorectal cancer, and prostate cancer, inhibition of these differentiation pathways in HNSCC may be an accessible and exciting avenue for novel therapeutics.
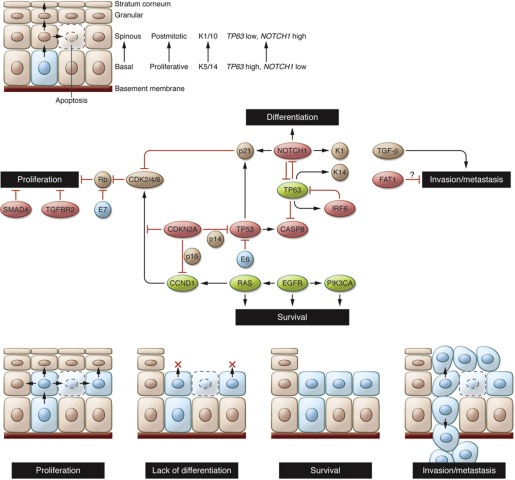
Sequencing data also have provided a rich array of data implicating loss-of-function mutations in additional pathways of differentiation in HNSCC, defining major new potential therapeutic targets. NOTCH1 loss-of-function mutations, for example, were noted in 11% to 19% of tumors, with another 11% to 14% containing NOTCH2 or NOTCH3 mutations (see Fig. 2 A). Interestingly, these same tumors had mutations in gene sets associated with differentiation, such as IRF6 and TP63, implying that these genes may act together with NOTCH1 to ultimately trigger the development of immature, dedifferentiated, highly proliferative basaloid cells. Additional mutations were identified in less well characterized genes such as SYNE1 and 2, which control nuclear polarity, and RIMS2 and piccolo presynaptic cytomatrix protein (PCLO), which regulate calcium sensing during terminal squamous cell differentiation. Thus, dysregulation of programs involving cellular differentiation appears to be a critical component of HNSCC tumor biology. In addition, inhibition of NOTCH activation has been associated with an increased risk of cutaneous SCC, with a recent phase III trial with the gamma-secretase inhibitor semagacestat halted due to an increased rate of cutaneous SCC in the treatment arm compared with placebo. Together, these data suggest that differentiation pathways may be a major regulator of HNSCC tumorigenesis, raising the possibility that dysregulation of differentiation identified in other SCCs of the lung, esophagus, and cervix may be relevant to head and neck biology.
Other tumor suppressor pathways also have been identified using unbiased approaches: FAT1, which has well-described roles in aberrant Wnt signaling, was mutated in 12% to 23% of tumors (see Fig. 2 A). Previous studies have shown that FAT1 encodes a cadherin-related protein that suppresses the nuclear localization of β-catenin and thereby inhibits proliferation. In addition, FAT1 appears to regulate cell migration and invasiveness (see Fig. 3 ), suggesting that there may be multiple effects of FAT1 mutations on HNSCC tumorigenesis. Additional mutations and deletions were identified in apoptosis-related genes (CASP8, DDX3X), histone methyltransferases (PRDM9, EZH2, NSD1), as well as Ajuba, a centrosomal protein that regulates cell division and vertebrate ciliogenesis in an EGFR-RAS-MAPK-dependent manner (see Fig. 2 A, C) ; however, further work is required to biologically characterize the mechanism and impact of these mutations on tumorigenesis. Nevertheless, identification of these mutations emphasizes the major role tumor suppressor pathways play in HNSCC pathogenesis.
Targeting tumor suppressor pathways is significantly more challenging than inhibiting oncogenic signaling, as it requires reactivation of tumor suppressor genes or their downstream effectors rather than simple chemical or biological inhibition. In this setting, the concept of synthetic lethality may prove useful for making therapeutic advances. Loss of a tumor suppressor gene may evoke unique susceptibilities to inhibition of a second gene or pathway that is normally not observed. Synthetic lethality leverages this principle that inhibition of two genes is lethal, whereas in contrast, inhibition of either gene alone is not.
There is now great interest in identifying the synthetic lethal partners of inactive genes, such as tumor suppressors, to help identify novel therapeutic targets. A recent data-driven computational approach for genome-wide identification of synthetic lethal interactions has been developed, with the ability to identify synthetic lethal partners of both oncogenes and tumor suppressors. This approach has been used to develop a network analysis of synthetic lethal interactions that predicts which genes are essential and likely to be efficient pharmaceutical targets. Although this analysis has not been completed in HNSCC, it will be critical to move forward swiftly with such an approach, taking advantage of genome-wide short hairpin RNA and small-interfering RNA–mediated drug sensitization and small molecular inhibitor screens. By characterizing the synthetic lethal genes across a broad array of head and neck tumors, one could identify the gene targets worthy of aggressive drug targeting. Such an approach leverages the bioinformatics power of network analyses, rather than trying to simply inhibit or activate a single gene target, an effect that may ultimately be escaped through evolution of the cancer cells themselves.
In more practical terms, McLornan and colleagues broadly characterized the pathways that may be nonoverlapping and unique to cancer cells; thus, targeting these aspects of cell biology may allow for selective targeting of cancer cells through synthetic lethality ( Box 1 ). For example, although DNA damage response pathways generally provide a “unified guard” against genomic instability, many malignancies have defects in aspects of DNA repair mechanisms. For example, mutations in the DNA repair gene MSH2 or MLH1 become synthetically lethal when combined with inhibitors of DNA polymerase due to the accumulation of double-strand breaks. Indeed, methotrexate has been shown to induce DNA damage in MSH2-mutant cells compared with wild type, which has been the basis for methotrexate trials in MSH2-deficient colorectal cancer.
Targeting oncogenic drivers
Exploiting DNA-repair or cell-cycle defects
Using new drug combinations derived from screen
Using altered drug timing and sequencing
Exploring the tumor-cell environment
Targeting the stroma
Exploiting the altered metabolome
Targeting the altered proteome
Exploiting nononcogene “addiction”
Targeting cells with p53 loss of function, which is mutated in 72% of HNSCC, may be one especially important context in which synthetic lethality approaches may prove valuable. In the context of p53 loss of function, cancer cells lose the normal mechanisms of p53-dependent G1-S cell-cycle arrest and become dependent on G2-M checkpoint arrest for DNA repair and survival. Thus, targeting G2-M checkpoint proteins can induce mitotic catastrophe and synthetic lethality in p53 loss-of-function cells. For example, inhibition of stress-activated p38 mitogen-activated protein kinase MAPKAP kinase 2 (MK2), ataxia telangiectasia mutated (ATM), and serum/glucocorticoid regulated kinase 2 (SGK2) or serine/threonine protein kinase 3 (PAK3) may sensitize tumor cells to chemotherapy (MK2, ATM) or induce autophagy (SGK2) or apoptosis (PAK3). Recent computational analyses have identified multiple candidate kinase genes that serve as synthetic lethal partners of p53 mutants, including polo-like kinase 1, cyclin-dependent kinase 1, and aurora kinase A. Within head and neck oncology, this approach has been put into practice with an RNAi kinome viability screen in p53 mutant HNSCC cells to identify oncogenes that may be targeted in this mutational context. Using this method to screen primary human HNSCC tumors, as well as tissue from murine models, several “critical survival kinases” were identified, including the Wee1-like protein kinase (WEE1). Small molecule inhibition of WEE1 using the compound MK-1775 revealed durable effects on HNSCC viability and apoptosis, while also potentiating the efficacy of cisplatin in a mouse xenograft model. This inhibitor is now part of a phase I clinical trial to determine whether it may be useful in combination with neoadjuvant weekly docetaxel and cisplatin before surgery in p53-mutant HNSCC. Exploring these approaches further is likely to yield additional novel therapeutics for loss-of-function mutations that have remained difficult to target.
Oncogene mutations are uncommon in head and neck squamous cell carcinoma with limited potential for targeted therapy in specific contexts
Unlike other malignancies, such as breast cancer or chronic myelogenous leukemia, which stand as examples of cancers driven by oncogenes (epidermal growth factor receptor [EGFR] and BCR-Abl, respectively) which can be inhibited with profound effects on clinical outcomes, HNSCC does not appear to demonstrate significant oncogene addiction. In vitro studies have identified a role for EGFR signaling in HNSCC, but sequencing analyses suggest only 15% of HPV– and 6% of HPV+ tumors contain mutations or amplification of EGFR (see Fig. 2 B). EGFR is a transmembrane tyrosine kinase receptor in the HER/erbB family of proteins that triggers Ras and PI3K signaling (see Fig. 2 C). In HNSCC, candidate sequencing studies have shown that EGFR is overexpressed most commonly through gene amplification and increased copy number, rather than activating mutations or truncation mutants such as EGFRvIII.
Based on the limited dependence of HNSCC on EGFR signaling, it is not surprising that inhibitors of EGFR have had variable success. EGFR overexpression appears predictive of poor clinical prognosis and resistance to radiation, with data suggesting improved overall survival when cetuximab, a monoclonal antibody against EGFR, is combined with radiation or chemotherapy. However, response to cetuximab does not correlate with the degree of overexpression and as a monotherapy, the benefits of cetuximab are limited to a 6% to 13% response rate. Similarly, the Radiation Therapy Oncology Group recently completed a Phase III trial exploring the effects of cetuximab in patients with stage III or IV HNSCC who were undergoing concurrent accelerated fractionated radiotherapy and cisplatin treatment. This group found no differences in patient outcomes (mortality, progression-free survival, overall survival, locoregional failure, or distant metastasis) with the addition of cetuximab. These findings suggest that other mechanisms may be activated upon EGFR inhibition or redundant activators of cell survival may limit treatment efficacy, consistent with whole-exome studies suggesting oncogenes have low mutant allele frequencies and rarely drive HNSCC (see Fig. 2 C). Thus, although there has been substantial interest in kinase inhibitors of EGFR in treating HNSCC, these agents have limited clinical impact in a significant portion of HNSCC tumors.
One exception for targeted therapy may be activating Ras or PI3K mutations, which occur at higher frequency in HPV+ cancers, offering a specific context in which targeted therapy may facilitate deintensification of chemoradiation. PI3K signaling is frequently altered in HNSCC through several mechanisms, including loss-of-function mutations in PTEN, which negatively regulates PI3K (40% of HNSCC) and activating mutations in PI3KCA (6%–11% of HNSCC). Recent data suggest that the PTEN gene may exhibit a gene dosage effect, with loss of a single allele promoting tumor growth. Interestingly, in the case PI3KCA, mutations may be associated with HPV+ OPSCC, raising the possibility that PI3K acts synergistically with HPV E6 and E7 proteins in this HNSCC subset.
Ras signaling may work in collaboration with PI3K activation or independently to promote HNSCC (see Fig. 2 C). Although KRAS is frequently mutated in other cancers, HNSCC is associated primarily with HRAS mutations, especially in patients with extensive tobacco exposure. Like PI3K, HRAS mutations are also associated with HPV+ tumors. However, Ras family members have proven recalcitrant to inhibition with therapeutic strategies primarily aimed at targeting downstream effectors.
Recent sequencing analyses have validated these in vitro observations identifying amplifications or mutations (specifically the exon 9 helical domain) of PI3K in 56% of HPV+ HNSCC and 34% of HPV– tumors (see Fig. 2 B). Activating mutations in HRAS were also described in 5% to 8% of HNSCC tumors (see Fig. 2 A). There are numerous ongoing trials evaluating small molecule inhibitors of PI3K. Such molecular inhibitors may be one exception in HNSCC, where oncogene targeting may prove valuable.
Human papilloma virus infection and integration alters tumor biology and triggers carcinogenesis by divergent biologic mechanisms than smoking and alcohol-related head and neck squamous cell carcinoma
More than a decade of research has made it clear that HPV+ and HPV– HNSCC are distinct entities, with unique etiology, patient demographics, pathophysiology, and clinical outcomes. We now know that HPV– cancers are those that are driven by traditional risk factors, such as smoking and alcohol, with carcinogenesis dependent on the acquisition of multiple epigenetic and genetic alterations yielding a premalignant progenitor that then undergoes additional alterations to become an invasive malignancy. This concept, known as field cancerization, posits that exposure of aerodigestive mucosa to alcohol and tobacco develops genetically distinct fields in which additional mutations may cause transformation. Consistent with this, Slaughter and Southwick made the early observation that 11.2% of HNSCC primaries present with a second primary. Similarly, Sidransky and colleagues examined 87 HNSCCs including analysis of preinvasive lesions using microsatellite analyses for loss of heterozygosity at 10 distinct loci. These analyses revealed progressive chromosomal loss when comparing benign hyperplasia to dysplasia to carcinoma in situ to invasive cancer, suggesting a common clonal progenitor and clonal expansion.
In contrast, HPV+ tumors are driven by HPV infection, usually by serotype16, with the integration of viral DNA into the host genome and the activation of specific and consistent molecular regulators, including p16 (INK4A) and viral proteins E6 and E7 (see Fig. 1 ). Cell lines transfected with p16 and the alternate transcript p14arf displayed markedly inhibited growth, arresting in G1consistent with a role for p16 in blocking the G1-S transition. Indeed, transfection of p16-INK4A adenovirus demonstrated a 96% reduction in proliferation of HNSCC cell lines and in vivo studies in nude mice showed a significant decline in xenograft tumor growth. More recent studies have linked p16-positive IHC (best defined as ≥70% cytoplasmic and nuclear staining) with HPV+ tumors and suggested that p16 overexpression be used as an independent factor to risk stratify OPSCC. Interestingly, epigenetic regulation of p16 through hypermethylation may also play an important role in predicting clinical prognosis and outcomes.
The biologic mechanisms explaining differences in clinical outcomes with HPV status are likely multifactorial. The absence of field cancerization certainly reduces the incidence of locoregional recurrence and second primaries, whereas the persistence of functional p53 may explain the improved response to chemotherapy and radiation. There is also growing evidence that tumor-immune interactions may explain the improved response of HPV+ tumors: HPV positivity is associated with a more substantial lymphocyte response and animal models suggest that immunocompetence is essential for complete tumor eradication.
More recent genome-wide studies of HPV+ and HPV– tumors reveal a clear divergence of these tumors at the genomic level. Compared with HPV– tumors, HPV+ HNSCC has lower rates of mutations and less frequent copy number alterations, indicating that there is less genomic instability in this cohort. In a recent study, Akagi and colleagues subsequently demonstrated that HPV integrants flank extensive regions of the host genome, resulting in amplifications and rearrangement. In addition, looping of HPV integrant-mediated replication leads to viral-host concatemers, thereby triggering oncogenesis. However, by DNA analysis there is no consistency in the site of HPV integration: interrogation of RNA transcripts demonstrated transcription across the viral-human integration locus with no recurrent genes identified, suggesting there are diverse mechanisms related to HPV integration, adding another potential source of intratumor and intertumor heterogeneity.
At the level of individual genes, recent whole-exome sequencing studies suggest that HPV+ tumors have infrequent mutations in p53 and CDKN2A in stark contrast to HPV– tumors where these genes are commonly altered. Interestingly, HPV+ tumors are distinguished by recurrent deletions and truncation mutations in tumor necrosis factor receptor–associated factor 3 (TRAF3), which has been implicated in innate and acquired viral response to Epstein-Barr virus, HPV, and human immunodeficiency virus. Loss of TRAF3 promotes aberrant nuclear factor kappa B (NFκB) signaling with diverse downstream effects on cytokine signaling and cell death. Thus, there is now unequivocal evidence that mechanistically separates HPV+ and HPV– HNSCC, clarifying the biological basis for the distinct clinical behavior of these distinct head and neck tumors.
Related posts:
 New Diseases and New Treatments—Head and Neck Cancer Updates
New Diseases and New Treatments—Head and Neck Cancer Updates
 Sequential and Concurrent Chemoradiation
Sequential and Concurrent Chemoradiation
 New Diseases and New Treatments—Head and Neck Cancer Updates
Molecular Aspects of Head and Neck Cancer Therapy
Transoral Endoscopic Head and Neck Surgery
Radiation Oncology—New Approaches in Squamous Cell Cancer of the Head and Neck
New Diseases and New Treatments—Head and Neck Cancer Updates
Molecular Aspects of Head and Neck Cancer Therapy
Transoral Endoscopic Head and Neck Surgery
Radiation Oncology—New Approaches in Squamous Cell Cancer of the Head and Neck
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


