Carcinogenic progression in the pancreas arises through a well-established stepwise accumulation of molecular aberrations from a normal cell to an invasive adenocarcinoma. Recent large-scale sequencing efforts have provided insight into novel driver genes as well as enriched core signaling pathways that underlie the inherent heterogeneity found in pancreatic cancer. By exploiting these genomic profiles, we may begin to provide new insights into patient stratification and therapeutic guidance. This review discusses the molecular landscape of pancreatic cancer and its role in tumor progression, clinical prognostication, and the development of novel therapeutic strategies.
Key points
- •
Carcinogenic progression of pancreatic cancer involves the stepwise accumulation of molecular aberrations from a normal cell to an invasive adenocarcinoma.
- •
Recent large-scale sequencing efforts have revealed novel driver genes and enriched core signaling pathways that abridge the inherent heterogeneity found in pancreatic cancer.
- •
By exploiting these genomic profiles, we may begin to provide new insights into patient stratification and therapeutic guidance.
Although rare (2% of cancer cases), pancreatic ductal adenocarcinoma (PDAC) is the fourth leading cause of cancer-related deaths in this country. In contrast with the decline of cancer-related deaths from other malignancies, the alarming increase in incidence of PDAC is projected to make it the second leading cause of cancer-related death by 2030. With a 5-year survival rate of only 4%, the lethality of this disease is attributed in part to the lack of early detection and a limited armamentarium of effective therapeutic strategies. The main oncogenic driver mutation, observed in greater than 90% of PDAC, KRAS (v-Kiras2 Kirsten rat sarcoma viral oncogene homolog) has also compounded its poor survival rate owing to its “undruggability,” although chemotherapeutic strategies may exist in certain targetable cases as discussed elsewhere in this paper.
Advances in next-generation sequencing technologies have allowed for a detailed insight into the genomic landscape of PDAC, to better understand how molecular alterations contribute to disease initiation and progression. In particular, dissecting the molecular events involved in the progression of PDAC from pancreatic intraepithelial neoplasia (PanINs) lesions to invasive carcinoma, are being achieved with possible implications to prognosis and targeted therapeutic approaches.
Multistep progression of pancreatic ductal adenocarcinoma
The progression of PDAC from a normal cell to an invasive adenocarcinoma involves the accumulation of inherited and/or acquired mutations throughout a span of up to approximately 20 years ( Fig. 1 ). This highlights the importance of exploiting this window of progression to develop new screening methods to provide curative surgical approaches. This progression involves the evolution of histologically recognized precursor lesions known as PanINs. As of the date of this publication, the categorization of PanIN is divided into low grade (PanIN-1A and 1B), intermediate (PanIN-2), and high grade (PanIN-3), although there is an emerging consensus in the clinical research community to move to a simplified 2-tiered classification of “low”-grade (PanIn-1 and -2) and “high”-grade PanINs (PanIN-3). This is based on observations that, although PanIN-1 and -2 lesions can be found even in the absence of cancer, PanIN-3 is almost never found without concomitant invasive neoplasia. Genetic alterations can be grouped into those that arise in precursor lesions and are usually, albeit not always, found in the concomitant PDAC, versus those that arise during subclonal evolution of the infiltrating carcinoma resulting in genetic heterogeneity.
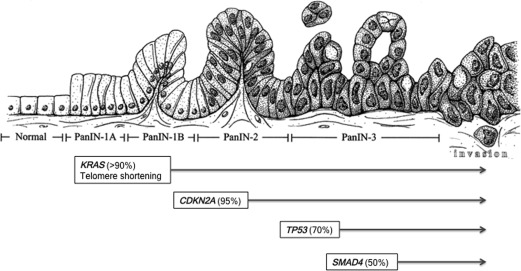
One of the earliest genetic events involved in PDAC pathogenesis is an activating point mutation in the KRAS oncogene, an oncogenic driver mutation found in more than 90% of all PDACs. Subsequent genetic aberrations include inactivation of tumor suppressor genes, including CDKN2A , TP53 , and SMAD4 , which encode for p16 INK4A , p53, and Smad4, respectively, and contribute to the histologic evolution of these precursor lesions. Together, these 4 alterations comprise the “big 4” in PDAC, although many other recurrent somatic mutations are found in lower frequencies (5%–10%) of cases, including those that afflict particular functional pathways in the cancer cell, such as DNA damage repair or chromatin regulation.
Among the population of noninvasive precursor lesions, it is also important to recognize intraductal papillary mucinous neoplasms (IPMNs) and mucinous cystic neoplasms. Although histologically distinct from the microscopic PanIN lesions, these cystic precursor lesions share similar diver mutations including point mutations in the KRAS oncogene and inactivating mutations in p53 and p16, with SMAD4 loss typically not being found in IPMNs. Among the unique drivers found in both IPMNs and mucinous cystic neoplasms are inactivating mutations in RNF43 , encoding for an ubiquitin ligase that has a role in WNT signaling inhibition. IPMNs also contain point mutations in the GNAS gene, which results in constitutively active guanine nucleotide-binding protein owing to loss of intrinsic GTPase activity preventing hydrolysis of guanosine 5′-triphosphate (GTP).
Multistep progression of pancreatic ductal adenocarcinoma
The progression of PDAC from a normal cell to an invasive adenocarcinoma involves the accumulation of inherited and/or acquired mutations throughout a span of up to approximately 20 years ( Fig. 1 ). This highlights the importance of exploiting this window of progression to develop new screening methods to provide curative surgical approaches. This progression involves the evolution of histologically recognized precursor lesions known as PanINs. As of the date of this publication, the categorization of PanIN is divided into low grade (PanIN-1A and 1B), intermediate (PanIN-2), and high grade (PanIN-3), although there is an emerging consensus in the clinical research community to move to a simplified 2-tiered classification of “low”-grade (PanIn-1 and -2) and “high”-grade PanINs (PanIN-3). This is based on observations that, although PanIN-1 and -2 lesions can be found even in the absence of cancer, PanIN-3 is almost never found without concomitant invasive neoplasia. Genetic alterations can be grouped into those that arise in precursor lesions and are usually, albeit not always, found in the concomitant PDAC, versus those that arise during subclonal evolution of the infiltrating carcinoma resulting in genetic heterogeneity.
One of the earliest genetic events involved in PDAC pathogenesis is an activating point mutation in the KRAS oncogene, an oncogenic driver mutation found in more than 90% of all PDACs. Subsequent genetic aberrations include inactivation of tumor suppressor genes, including CDKN2A , TP53 , and SMAD4 , which encode for p16 INK4A , p53, and Smad4, respectively, and contribute to the histologic evolution of these precursor lesions. Together, these 4 alterations comprise the “big 4” in PDAC, although many other recurrent somatic mutations are found in lower frequencies (5%–10%) of cases, including those that afflict particular functional pathways in the cancer cell, such as DNA damage repair or chromatin regulation.
Among the population of noninvasive precursor lesions, it is also important to recognize intraductal papillary mucinous neoplasms (IPMNs) and mucinous cystic neoplasms. Although histologically distinct from the microscopic PanIN lesions, these cystic precursor lesions share similar diver mutations including point mutations in the KRAS oncogene and inactivating mutations in p53 and p16, with SMAD4 loss typically not being found in IPMNs. Among the unique drivers found in both IPMNs and mucinous cystic neoplasms are inactivating mutations in RNF43 , encoding for an ubiquitin ligase that has a role in WNT signaling inhibition. IPMNs also contain point mutations in the GNAS gene, which results in constitutively active guanine nucleotide-binding protein owing to loss of intrinsic GTPase activity preventing hydrolysis of guanosine 5′-triphosphate (GTP).
KRAS
Under physiologic conditions, activation of Ras protein is induced through growth factor receptor signaling, which promotes Ras activity through transitory binding to GTP. This results in the interaction of Ras with a variety of downstream effectors that govern proliferation, cell division, survival, and gene expression such as the RAF-mitogen-activated protein kinase and phosphoinositide 3-kinase pathways. Through an intrinsic GTPase mechanism and GTPase-activating proteins, Ras can then hydrolyze GTP to GDP to inactivate itself. It is this intrinsic GTPase activity that is altered in the activating point mutation of KRAS, which results in an inability to hydrolyze GTP, allowing for a constitutively active protein that no longer relies on external stimuli. The most common “hotspot” mutation in the KRAS oncogene occurs at codon 12, followed by codon 13, and less frequently codon 61; emerging genomic data suggest that the specific codon involved might have an impact on disease prognosis, underscoring differences in Ras function.
Telomere shortening
Telomeres are repetitive nucleoprotein complexes found at the end chromosomes that have a role in genomic stability by protecting against chromosomal degradation and chromosome end fusion. In PDAC, shortened telomere lengths that potentially lead to chromosomal abnormalities can be detected in early lesions such as IPMNs and are nearly universally in all PanINs. Genomic instability of this kind typically leads to cell death unless cells are able to inactivate tumor suppressor mechanisms as described elsewhere in this paper.
CDKN2A
The most commonly mutated tumor suppressor in PDAC (∼95%) is an inactivation of cyclin-dependent kinase inhibitor 2A gene ( CDKN2A ), encoding for the cell cycle checkpoint protein, p16 INK4A . Inactivation of CDKN2A in PDAC can occur via several different mechanisms, including mutation, genomic deletions, and promoter hypermethylation, resulting in epigenetic silencing. The encoded protein p16 INK4A functions as a cyclin-dependent kinase inhibitor, specifically of CDK4 and CDK6, thereby preventing the phosphorylation of the retinoblastoma protein and blocking G1-S transition. Loss of this protein thus results in unregulated cell cycle transition.
TP53
Aberrations of TP53 , which encodes for p53, are typically a later event in the multiprogression of PDAC and is mutated in up to 70% of tumors, often arising in PanIN-3 lesions. As the master guardian of the genome, p53 is involved in cell cycle arrest, DNA repair, blocking of angiogenesis, and induction of apoptosis in response to DNA damage or environmental stressors. Loss of this protein allows for DNA damage and external stressors to go unchecked, thereby promoting genomic instability and aberrant proliferation.
SMAD4
SMAD4 (DPC4, SMAD family member 4 gene), which encodes for the Smad4 protein, is inactivated in approximately 50% of PDACs as a late event in its progression (PanIN-3 – carcinoma). As a downstream effector of transforming growth factor-β, loss of SMAD4 activity leads to tumor promotion by relieving the growth inhibitory effect of transforming growth factor-β signaling.
Clinical relevance of core pancreatic ductal adenocarcinoma mutations
Although pancreatic cancers have been shown to harbor an average of 63 genetic mutations, the genomic landscape of PDAC is faithfully represented by these 4 genomic mutations (KRAS, CDKN2A, TP53, and SMAD4). The high degree of mutational concordance at these 4 loci between primary and metastatic sites of individual cancers suggests these are so-called founder mutations. This term describes a mutation present in all samples from a single patient, thus sharing a common ancestor, which likely originated during PanIN progression. Of note, PDACs continue to accumulate genetic alterations through subclonal evolution throughout their natural history, the vast majority of which are so-called passenger mutations that have little functional impact on progression, whereas a minor fraction are so-called progressor mutations, and do have a deleterious consequence on disease progression. In any case, a variable combination of the “big 4” is altered in most PDAC cases, with nearly all cancers showing at least KRAS mutations in conjunction with one or more of the 3 tumor suppressors. Yachida and colleagues describe correlations of the status of these 4 genes to disease progression, metastatic failure, and overall survival, with patients with 3 to 4 of these mutated driver genes demonstrating worse overall survival. When looking at the genes individually, there is no significant difference in outcome in patients with KRAS and CDKN2A mutations, but TP53 and SMAD4 mutations were evidently associated with widespread metastatic disease and worse clinical outcomes. In particular, SMAD4 status in PDAC (measured using immunohistochemical expression for the Smad4 protein) is being used to provide guidance toward tailoring treatment with systemic chemotherapy, as patients with Smad4-null tumors are most likely to develop widely metastatic disease.
Germline variants
With estimates of 10% of PDAC patients having a family history of the disease, elucidation of the PDAC genome has also contributed toward risk assessment and early detection in the context of familial pancreatic cancers. Among hereditary pancreatic cancer susceptibility genes, STK11/LKB1 , which is associated with Peutz–Jegher syndrome, is correlated with one of the highest risks of familial pancreatic cancers with approximately 132 greater relative lifetime risk. PRSS1 and SPINK1 germline mutations are seen in hereditary pancreatitis families with a 50× to 80× relative lifetime risk, or 30% to 44% risk, of developing PDAC. P16/CDKN2A germline mutations, are associated with familial atypical multiple mole melanoma syndrome, which entails a 38× increased risk (17% lifetime risk) of developing PDAC. Additional germline variants associated with increased PDAC risk are clustered into defects of DNA repair pathways. This includes members of the Fanconi anemia pathway such as FANC-C, FANC-G, and PALB2, whose encoded proteins interact with that of BRCA2, and are associated with young onset pancreatic cancer. BRCA1/BRCA2 mutations, which are associated with familial breast and ovarian cancers, have a 3.5× to 10× estimated relative risk. Lynch syndrome, caused by mutations in DNA mismatch repair genes, MLH1, MSH2, MSH6, or PMS2, have an estimated 3.68% lifetime risk. Patients with ataxia telangiectasia mutated germline mutations have also demonstrated a predisposition for PDAC. Further elucidation of additional genes associated with familial PDAC may have implications for risk assessment and surveillance in affected family members. Detection of these germline mutations in patients is also important in the context of therapy as a way to exploit synthetic lethal interactions in the case of DNA repair pathways as described elsewhere in this paper.
Core signaling pathways in pancreatic cancer
Large-scale sequencing efforts have uncovered novel mutated genes in PDAC, as well as revealing multiple core signaling pathways that are affected throughout its carcinogenesis. In 2008, Jones and colleagues performed polymerase chain reaction–based exome sequencing of primary and metastatic tumors. Their data supported the role of the 4 main genetic drivers in pancreatic cancer, KRAS, CDKN2A, TP53, SMAD4, and identified genetic alterations in many other critical pathways recognized as “hallmarks of cancer,” at least some of which seem to have a prognostic influence in subsequent studies. The core “hallmarks of pancreatic cancer” pathways that seem to be targeted in PDAC are highlighted in Table 1 with some salient examples of genes mutated in each pathway.
Related posts:
 Role of Endoscopic Ultrasonography and Endoscopic Retrograde Cholangiopancreatography in the Clinical Assessment of Pancreatic Neoplasms
Role of Endoscopic Ultrasonography and Endoscopic Retrograde Cholangiopancreatography in the Clinical Assessment of Pancreatic Neoplasms
 Minimally Invasive Approaches to Pancreatic Surgery
Adjuvant and Neoadjuvant Therapy for Pancreatic Cancer
Clinical Presentation and Diagnosis of Pancreatic Neuroendocrine Tumors
State-of-the-art Imaging of Pancreatic Neuroendocrine Tumors
Surgical Management of Pancreatic Neuroendocrine Tumors
Minimally Invasive Approaches to Pancreatic Surgery
Adjuvant and Neoadjuvant Therapy for Pancreatic Cancer
Clinical Presentation and Diagnosis of Pancreatic Neuroendocrine Tumors
State-of-the-art Imaging of Pancreatic Neuroendocrine Tumors
Surgical Management of Pancreatic Neuroendocrine Tumors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


