Fig. 1
Cartoon outlining key steps during mouse pituitary development. Rathke’s pouch (RP) forms at 9.5 dpc (days post coitum) as a thickening of the oral ectoderm (OE) at the boundary between OE and pharyngeal endoderm (PE) that invaginates toward the ventral diencephalon (DI). The epithelium of RP contains the undifferentiated progenitors that give rise to all hormone-producing cells by the end of gestation. By 11.5 dpc, the invaginating RP pinches off from the oral ectoderm and establishes contact with the infundibulum (INF), a recess of the floor of the DI. RP progenitors proliferate rapidly from 9.5 dpc and the progeny migrate ventrally and initiate differentiation to populate the primordium of the anterior lobe (AL) of the pituitary. The presumptive intermediate lobe (IL) is indicated. At 18.5 dpc, just prior to birth, the pituitary comprises the posterior pituitary (PP), of neural origin, and the anterior pituitary (AP), derived from the anterior and intermediate lobes. The pituitary is located between the hypothalamus (Hyp) and the basisphenoid cartilage (BS). Reproduced with permission from Wiley (Martinez-Barbera and Andoniadou (2016) Stem Cells)
Normal morphogenesis and differentiation of the pituitary gland is regulated by the activity of several transcription factors and signalling pathways [2–4]. One of these pathways is WNT/β-catenin, a major regulator of several cellular processes during organogenesis and adulthood [5, 6]. The activity of the WNT/β-catenin pathway is controlled by the stability of β-catenin, a transcriptional activator lacking a DNA-binding domain. In the absence of WNT ligands, β-catenin is phosphorylated by a destruction complex and degraded rapidly via the ubiquitination pathway, preventing its stabilisation and the activation of the WNT cascade. Binding of WNT ligands to their receptors disrupts the destruction complex preventing the phosphorylation of β-catenin and its subsequent degradation. Stabilised β-catenin can translocate into the nucleus and interact with DNA-binding transcription factors of the TCF/LEF family to activate the expression of target genes (Fig. 2).
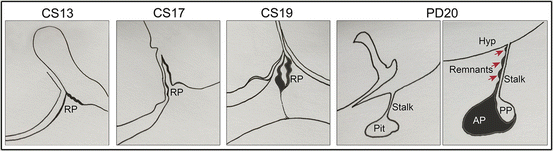
Fig. 2
Cartoon outlining key steps during human pituitary development. By Carnegie Stage (CS) 13, invagination of Rathke’s pouch (RP) has initiated. This makes contact with the ventral diencephalon from CS17. By CS19 the rudimentary pouch has formed and is detaching from the oral epithelium. The postnatal pituitary gland (Pit), presented here at postnatal day (PD) 20, is connected to the hypothalamus (Hyp) via the pituitary stalk (Stalk), with axons terminating in the posterior pituitary (PP). A distinct feature of the human pituitary compared to mouse is the longer pituitary stalk, on which are found remnants of anterior pituitary (AP) tissue
In humans, the morphogenesis of the HP axis shares a common embryonic origin with mice and the same genes and pathways are deployed during its development. However, there are some species-specific differences that are important to keep in mind. In both human and mouse embryos, the precursors of the anterior pituitary in Rathke’s pouch are initially in contact with the overlying hypothalamic primordium and their descendants must migrate into the sellar region to form the anterior pituitary. The primordium of the posterior pituitary (i.e. infundibulum) is initially embedded within the hypothalamus and moves downwards to its final position adjacent to the anterior pituitary within the sella turcica (Fig. 3). However, the pituitary stalk is markedly longer in human embryos and pituitary cells must migrate further during morphogenesis [7]. Developmental defects in these morphogenetic movements lead to Pituitary Stalk Interruption Syndrome (PSIS) in humans, which is characterised by the presence of a thin or absent pituitary stalk, commonly associated with a hypoplastic or aplastic anterior pituitary or an ectopic posterior pituitary (e.g. attached to the floor of the 3rd ventricle, optic chiasm or median eminence) [8, 9]. Another peculiarity of the human HP axis, a feature not reported in mice, is the presence of pituitary tissue along the dorso-ventral axis of the stalk. These pituitary ‘nests’ were initially observed by Jakob Erdheim in 1904 [10], suggesting that remnants of RP could be trapped during normal development of the HP axis, a finding that could have implications in the location of pituitary tumours within the sella region, such as whether supra- or intra-cellar.
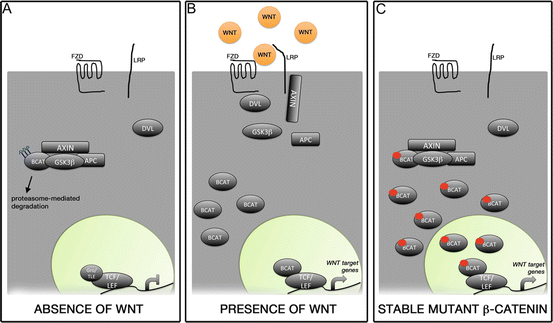
Fig. 3
WNT/β-catenin signalling pathway. (a) In the absence of WNT ligands, β-catenin is in a destruction complex formed by AXIN, APC and GSK3β and phosphorylated in specific residues encoded by exon 3 of CTNNB1. This results in rapid degradation of β-catenin following ubiquitination, and consequently, the repression of target genes brought about by the interaction of TCF/LEF1 with co-repressors (e.g. groucho (Gro)/TLE family members). (b) Binding of WNT ligands to Frizzled (FZD) receptors and co-receptors LRP leads to the recruitment of dishevelled (DVL) and AXIN to the cytoplasmic membrane. This causes the disruption of the destruction complex, releasing β-catenin, which cannot be efficiently phosphorylated and degraded. The stabilisation of β-catenin results in increased cytoplasmic protein levels and eventually nuclear translocation, where its interaction with TCF/LEF1 factors mediates the expression of pathway target genes. (c) Expression of a stable form of β-catenin lacking the amino acids encoded by exon 3 of CTNNB1 (small red circle in BCAT) results in protein stabilization, since mutant β-catenin cannot be targeted for degradation by phosphorylation at these residues. This causes the activation of the WNT/β-catenin pathway in a cell-autonomous manner, even in the complete absence of WNT ligands. Reproduced with permission from Wiley (Martinez-Barbera (2015) Neuropathol Appl Neurobiol)
Pathology of Human ACP
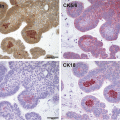
Human ACPs are cystic tumours that develop within the sellar region, frequently compromising the hypothalamus, pituitary gland, pituitary stalk and optic chiasm. The histopathology of these tumours is characterized by the formation of a peripheral basal cell layer of palisading epithelium, loose aggregates of stellate cells, nodules containing anucleated ‘ghost cells’ and ‘wet keratin’, large areas of regressive changes, i.e. hemosiderin deposits, cholesterol clefts, multinucleated foreign-body giant cells, inflammation and calcifications (Fig. 4a) [11–14]. Human ACPs are usually benign tumours but there have been rare cases of malignant ACP, perhaps linked to the use of radiotherapy [15–17]. In these malignant neoplasms, P53 is over-expressed [18], alluding to the possibility that other mutations may have been induced as a result of the irradiation thus generating more aggressive tumours.
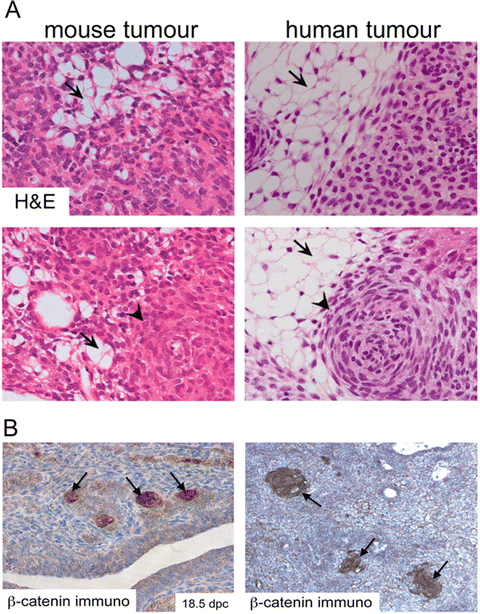
Fig. 4
Histomorphological features of mouse and human ACP. (a) Haematoxylin and eosin staining of mouse and human tumours showing the presence of microcystic changes (stellate reticulum; arrows) and whorl-like nodular structures (cell clusters; arrowheads). (b) Immunohistochemistry with a specific anti-β-catenin antibody showing the presence of cell clusters with nucleo-cytoplasmic accumulation of β-catenin. Reproduced with permission from PNAS (Gaston-Massuet et al. (2011) Proc Natl Acad Sci USA)
Mutations in CTNNB1 have been identified in the majority of the human ACP samples analysed [19–24]. These are mostly restricted to exon 3 and result in the substitution or deletion of the critical regulatory amino acids that control stabilisation of the protein, predicted to lead to its nucleo-cytoplasmic accumulation and over-activation of the WNT/β-catenin pathway [25–29]. Global expression analyses have identified several genes/pathways that are dysregulated in human ACP, some of which could be targeted using drugs that are currently approved [30, 31]. However, little is known about the function of such pathways in the pathogenesis of these neoplasias, e.g. whether these may be pro- or anti-tumourigenic.
A histopathological hallmark of human ACP is the presence of cells showing nucleo-cytoplasmic accumulation of β-catenin, commonly grouped forming whirl-like structures close to the invasive front (hereby referred to as ‘cell clusters’) [20, 21, 24, 32, 33] (Fig. 4b). These are clearly a minority of cells and are distributed evenly throughout the tumour. Clusters accumulating β-catenin activate the WNT/β-catenin pathway, as evidenced by the elevated expression of pathway targets such as AXIN2, LEF1 and BMP4 [26, 28, 32]. The presence of clusters is a histological hallmark that differentiates human ACP from any other tumour of the sellar region, including papillary craniopharyngioma [27].
Over-Activation of the WNT/β-Catenin Pathway Underlies the Molecular Aetiology of Human and Mouse ACP
Genetic studies in mice have demonstrated the requirement of the WNT/β-catenin pathway for normal pituitary morphogenesis and differentiation in both mice and humans [36]. WNT/β-catenin is necessary for terminal differentiation of hormone-producing cells at late stages of pituitary development, whilst at early stages it must be regulated for normal proliferation of Rathke’s pouch progenitors, e.g. if not inhibited results in overproliferation [37–42]. Supporting this notion, the up-regulation of the WNT/β-catenin pathway in Rathke’s pouch (RP) progenitors leads to dramatic pituitary hyperplasia, ectopic pituitary tissue in the oropharyngeal cavity and perinatal death in 80% of pups. In addition, somatotroph, lactotroph and thyrotroph differentiation is impaired [26]. This genetically modified mouse model (Hesx1 Cre/+ ;Ctnnb1 lox(ex3)/+ ) harbours a mutated form of β-catenin that is transcriptionally active, but lacks the amino acids encoded by exon 3 of Ctnnb1 (the gene encoding β-catenin), preventing its phosphorylation and subsequent degradation, leading to the over-activation of the WNT/β-catenin pathway, akin to human mutations in exon 3 [43].
An interesting observation of the previously described embryonic mouse line is that pups which survive weaning (around 20%) show variable growth defects, consistent with an impairment in somatotroph differentiation, and fully penetrant pituitary tumours [26]. Histopathological analysis revealed characteristic hallmarks of human ACP, such as large cysts, cell clusters with nucleo-cytoplasmic accumulation of β-catenin and expression of Axin2, Lef1 and Bmp4, indicating the activation of the WNT/β-catenin pathway [25–27]. These neoplastic cells did not express markers of proliferation or pituitary hormones and were negative for synaptophysin, a marker of neurons and endocrine cells. Therefore, cluster cells in mouse and human ACP have exited the cell cycle, are undifferentiated and, possibly not of either neural or endocrine origin. However, contrary to the human histopathology, mouse ACP did not display wet keratin or calcification, suggesting species-specific differences between mouse and human ACP. Critically, human ACP develop over years, but mouse ACP develop over a period of weeks. Despite these differences, the mouse tumours are more similar to human ACP than to any other malformation or tumour of the sellar region including pituitary adenoma, Rathke’s cleft cysts and even the papillary type of craniopharyngioma. Together, the human and mouse studies have provided robust evidence for a role of CTNNB1 mutations and the over-activation of the WNT/β-catenin pathway in the molecular aetiology of ACP.
Childhood-Onset ACP is Likely to be a Tumour of Embryonic Origin
The molecular and genetic studies using the embryonic ACP mouse model revealed two key findings, of relevance to the pathogenesis of human ACP, and in particular childhood-onset ACP. Analyses of mouse embryos at different stages of development established these form β-catenin-accumulating cell clusters with a similar gene expression profile to the human clusters and that these are established by the end of gestation [26]. Second, that in these mice, tumours only form when oncogenic β-catenin is expressed in Rathke’s pouch undifferentiated precursors, but not when committed progenitors or fully differentiated hormone-producing cells are targeted [26].
Are these findings applicable to the human tumours? The similarities between the mouse and human clusters are remarkable, not only morphologically but also at the molecular level, strongly suggesting that they represent equivalent structures in both species. In fact, whole transcriptome gene expression profiling of the mouse cluster cells revealed numerous genes/pathways that were dysregulated in mouse and also in human tumours [30, 31, 44–46]. The possibility that ACP patients may have cell clusters in the pituitary gland is supported by clinical evidence. In a retrospective analysis of 90 children diagnosed with ACP (median age at diagnosis 8.3 years), Müller and colleagues reported that a significant decrease in height score was noted at 10–12 months of age and persisted until the diagnosis of ACP [47]. Taken into consideration that somatotrophs are clearly defective in the embryonic ACP model by the end of gestation and postnatally result in dwarfism [26], it is prudent to suggest that clusters may develop during gestation of human patients. In fact, ACP was recently diagnosed in a human foetus at 18 weeks [48].
Taking into account the previously described anatomical differences in the length of the pituitary stalk as well as the more pronounced cephalic flexure of the developing human embryo [49], it is possible that tumour epithelial cells (i.e. destined to be cluster cells) could be trapped in the suprasellar region near the hypothalamus. These could eventually give rise to a tumour with little or no sellar component at all. This is compatible with the remnants of pituitary tissue identified along the dorso-ventral axis of the human pituitary stalk. It could be speculated that the position of the tumour-initiating cell targeted with the CTNNB1 mutation could determine the location of the ACP tumour in relation to the sella. Therefore, it is possible that the distinct behaviour of human ACP, for instance regarding its infiltrative behaviour, could be related to the position of the tumour initiating cells rather than to specific differences in its genetic/epigenetic landscape. Further molecular analyses will clarify this hypothesis.
Pituitary Stem Cells are Present in Mouse and Human Pituitaries
In recent years, solid evidence has gathered for the existence of a tissue-specific population of undifferentiated progenitors or stem cells within the hormone-secreting anterior pituitary gland of the mouse (pituitary stem cells, PSCs). These long-lived, undifferentiated cells have the capacity to directly commit or generate daughter cells of all cell lineages.
The transcription factor SOX2 is highly associated with pluripotency in the early embryo, in embryonic stem (ES) cells and induced pluripotent stem (iPS) cells, as well as the stem cell state in multiple embryonic and adult tissues. In the pituitary it was shown to define a population, which exclusively contains cells capable of both adherent and non-adherent clonogenic colony formation [44, 50]. In rodents at postnatal stages, SOX2-positive cells show a high degree of overlap with other proposed stem cell markers in vivo, such as PROP1 [51, 52], PRX1/2 [53] and SOX9 [54]. SOX2-expressing cells with similar clonogenic potential have been identified in human pituitaries [55].
Although SOX2-positive cells do not express hormones and SOX2 expression does not generally overlap with expression of the committed progenitor markers, the population has a heterogeneous nature in terms of properties, where only a low proportion (2.5–5%) are shown to be capable of in vitro self-renewal. Purifying pituitary cells based on S100β expression using S100β-GFP transgenic animals enriches in vitro colony formation, a property lying exclusively within the SOX2-positive compartment [56], hence suggesting that the overlapping population is likely to be enriched in stem cells and the strong association of S100β with multiple proposed pituitary stem cell populations makes this protein a good candidate marker. Indeed, there is significant overlap between SOX2 and S100β positivity [50, 56]. On its own, S100β marks multiple populations, displaying expression in the SOX2-positive marginal zone region, in the morphologically distinct folliculostellate cells and reported to be expressed in pituicytes. Multiple functions have been put forward for S100β-positive folliculostellate cells, reviewed comprehensively elsewhere [57], including providing structural support and enabling communication within the pituitary thus facilitating pulsatile hormone secretion [58, 59]. A further attribute of these cells is the production of paracrine signals that contribute to the regulation of surrounding endocrine cells. For example, synthesis and secretion of FSH in gonadotrophs is mediated by activin, and regulated by the activin-binding protein follistatin, both of which are produced by folliculostellate cells [60]. Numerous other paracrine factors secreted by these cells have been identified, including interleukin-6 (IL-6), leukemia inhibitory factor (LIF), vascular endothelial growth factor (VEGF) and basic fibroblastic growth factor (bFGF), all of which contribute to pituitary function [61–63].
Contribution of Stem Cells in the Long-Term Maintenance of the Anterior Pituitary
Despite a plethora of identified markers, until recently there was no evidence to support that pituitary stem cells function as such in vivo. This changed with the generation of two similar inducible mouse strains expressing CreERT2 under the regulation of the Sox2 promoter, generated by the Hochedlinger and Martinez-Barbera/Pevny labs [56, 64]. In these, Cre recombinase is expressed by cells expressing Sox2, but will not be active in the nucleus until the administration of tamoxifen, allowing temporal control of recombinase action. Although the expression construct of these two strains is not identical, the expression of CreERT2 faithfully reproduces that of Sox2 in both cases. Sox2-expressing cells were found to give rise to all committed progenitor cell types (PIT1 (POU1F1), TPIT (TBX19), SF1 (NR5A1)), and thus all hormone-secreting cells types of the anterior lobe (GH, PRL, TSH, ACTH, LH/FSH). In addition, SOX2+ stem cells were shown to compose a long-lived population, either persisting long-term or maintained as a self-renewing pool of stem cells. Similar results were observed using a mouse expressing CreERT2 under the control of the Sox9 gene (Sox9-CreERT2 mouse line) [54]. This is not surprising as SOX2- and SOX9-expressing cells represent the same population. The above experiments demonstrate the presence of a long-lived population that retains pituitary stem cell properties throughout normal life.
Paracrine Contribution of Stem Cells to Tumourigenesis
As previously noted, mouse ACP form only when embryonic progenitors are targeted. Confirming this notion, expression of oncogenic β-catenin specifically in SOX2+ stem cells of the embryonic or adult pituitary (Sox2 CreERT2/+ ;Ctnnb1 lox(ex3)/+ mouse model) also result in tumours that are similar to human ACP [56]. These mouse tumours contain cell clusters showing nucleo-cytoplasmic β-catenin and are undifferentiated (i.e. do not express any hormone), as the human tumours. The postnatal mouse model has been used to trace the descendant cells of pituitary stem cells (PSCs) which are mutated, a technique referred to as ‘genetic tracing’ [65]. SOX2+ cells are targeted to simultaneously express oncogenic β-catenin and Yellow Fluorescent Protein (YFP); the former is the oncogenic stimulus and the latter allows identification of all the daughter cells derived from the mutated SOX2+ PSCs. These experiments have revealed that the typical cell clusters derive from SOX2+ cells, but intriguingly, the bulk of the tumour mass does not. In other words, mutated SOX2 + cells are not transformed into tumour-propagating cancer stem cells (CSCs) by oncogenic β-catenin; instead, mutated SOX2+ PSCs generate cell clusters that have the capacity to induce tumours in a paracrine fashion and the tumour cells are not derived from these SOX2+ cells [56] (Fig. 5). Strengthening the notion of this paracrine action, cluster cells express a vast array of growth factors, chemokines and cytokines and act as signalling centres, possibly changing the tumour microenvironment and facilitating tumourigenesis [44, 56]. Of relevance, expression of several of these cytokines and growth factors have been shown to play a role in normal pituitary physiology as well as in pituitary adenoma, a more prevalent pituitary tumour in humans [61, 66, 67].
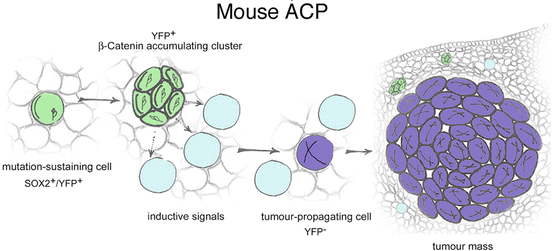
Fig. 5




Paracrine model of involvement of pituitary stem cells in tumourigenesis. When targeted to express oncogenic β-catenin, SOX2+ cells (green) accumulate nucleo-cytoplasmic β-catenin, proliferate transiently and stop dividing to form distinct clusters. Clusters secrete signals to the surrounding cells that may induce transformation and proliferation, resulting in tumour growth originating from a cell that is not derived from the targeted SOX2+ stem cell. Reproduced with permission from Elsevier (Andoniadou et al. (2013) Cell Stem Cell)
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
