Fig. 7.1
A model of PCa progression. PCa starts as a localized tumor. In its natural progression, it typically metastasizes to the lymph nodes and then the bone, producing bone-forming metastases. Most PCas respond to androgen ablation therapy, but relapse usually occurs, and bone is the primary site of progression
A more comprehensive understanding of PCa resulted in clinical division of the disease into subgroups having various patterns of progression to metastasis [13]. The pathologic classification of PCa is defined using the Gleason sum score (UICC), which is based on morphologic criteria [14]. Researchers categorized the clinical course of PCa from diagnosis to death as a series of clinical stages or treatment statuses (e.g., extent of the local disease, hormonal status, the absence or presence of detectable metastases on an imaging study). Prostate-specific antigen level is widely used to monitor disease progression and response to treatment. Although these clinical stages or treatment statuses are currently used to establish therapeutic objectives and outcomes, investigators have proposed a new molecular classification of PCa that incorporates androgen receptor level, oncogenes/tumor suppressors, and the tumor-bone microenvironment in the disease model. This proposed classification may facilitate the implementation of current and emerging therapies [15].
The cascade of events that lead to metastatic dissemination starts long before a tumor is clinically detected at a distant site [4, 16]. Metastatic dissemination possibly continues throughout the progression of the disease. Therefore, precise understanding of the molecular mechanism underlying each step in the metastatic process requires longitudinal analyses of human cancer progression in addition to laboratory-based studies. Some such studies have demonstrated the anarchic evolution of metastases, which in some cases caused local recurrence, whereas others had cross-metastatic site seeding [4, 16, 17]. In fact, one study demonstrated the presence of metastatic and primary tumor clones in blood even years after removal of the primary tumor, supporting the concept the anarchic evolution of metastases [16].
Solid tumors are typically heterogeneous, and studies have suggested the existence of clonal evolution of metastases as well as genetic and epigenetic diversity of primary and metastatic clones [4, 18]. This clonal diversity may be reduced or enhanced at both primary and metastatic sites by systemic treatment.
In this chapter, we primarily discuss epithelial-derived cancers with special emphasis on modeling the metastasis of PCa. PCa is the second leading cause of cancer deaths among American men. PCa cells are particularly difficult to grow on two-dimensional platforms, which are evidenced by the paucity of PCa cell lines available for study. Thus, PDXs are not only clinically relevant but, in the case of PCa, essential for PCa research given the paucity of cancer cell lines.
PCa PDX Models
In 1996, with the support of the Prostate Cancer Foundation, we established the PCa PDX program in the Department of Genitourinary Medical Oncology at MD Anderson. This program has the goal of developing PDXs from annotated tissue samples obtained from men undergoing radical prostatectomy, cystoprostatectomy/pelvic exenteration, or resection/biopsy analysis of metastatic lesions. PDXs are derived from advanced therapy-naïve prostate tumors or prostate tumors resistant to various therapies. By generating PDXs from different areas of the same tumor, we have developed models of PCa heterogeneity. PDXs developed in our facility include adenocarcinomas derived from therapy-naïve and therapy-resistant primary tumors and metastases [19–21]. Atypical clinical and histopathological variants PCa are also available [21, 22]. To date, the program has processed tumor samples derived from more than 270 patients with PCa for PDX development and established two PCa cell lines (MDA PCa 2a and 2b) and PCa PDXs from more than 76 donors. PDXs developed in our program are derived from patients with advanced PCa. We derived PCa PDXs from tumors in the prostate; areas of direct tumor extension to adjacent organs, the bone, lymph nodes, the liver, the thyroid, a testis, the adrenal gland, the brain, and unusual sites (e.g., skin, chest wall, soft tissue); ascites; and pleural effusions. PDXs are identified by the prefix MDA PCa followed by a number that is unique to the donor tumor, tumor site, and procedure date (e.g., MDA PCa 144). Because this is an ongoing program, the clinical and molecular evolution of PCa as a result of the development of resistance to new and upcoming therapies will be reflected in our dynamic repository. Because this PDX repository is dynamic, the number of developed PDXs provided above will change over time. The use of these PCa PDXs by investigators has been described in various reports of their research [19–44].
Also, to be able to study and understand the heterogeneity of PCa, when the tumor of origin is large, we submit human PCa tissue samples from different areas of the same tumor for PDX development, which are established as independent PDXs and independently identified according to a unique suffix, such as MDA PCa 144-2 and MDA PCa 144-4.
The PCa PDX program operates within a highly integrated network of physicians, scientists, staff, and resources in the Tissue Biospecimen and Pathology Resource at MD Anderson. These individuals include urologists, oncologists, pathologists, staff of the Department of Diagnostic Radiology (when tissue samples are obtained using image-guided biopsies), and staff who provide regulatory and compliance support for archived tissue and blood sample requests, consent validation, protocol submissions to the MD Anderson Institutional Review Board, material transfer agreements, data management, and logistic issues concerning sample shipments. This team ensures that complete patient information is captured for comparison with the derived PDXs.
Patient-Derived Xenografts as Models for Studying Metastasis
In recent years, the scientific community has recognized that patient-derived xenografts (PDXs) are far superior to cell lines for studying mechanisms of response of cancer to treatment with US Food and Drug Administration-approved and investigational agents and in discerning mechanisms of treatment resistance of human cancer. Furthermore, specific pathways involved in the metastatic process may be better reflected by early-passage PDXs, although cell lines are still useful in mechanistic studies.
Selection of approaches to modeling metastases using PDXs should be based on the specific pattern of progression of the disease of interest while taking into consideration the stages of the metastatic process that are to be modeled as well as heterogeneity and clonal evolution. For example, when studying the growth of PCa cells in the bone, in addition to using bone metastasis-derived PDXs, considering whether the process to be modeled would be influenced by the treatment status of the patient may be beneficial.
Modeling Specific Steps Involved in Metastasis
Cell Migration and Invasion
As described previously [1], migration and invasion are the initial events required for a solid tumor to spread and disseminate throughout the body. Cell migration is a multistep process; the cell motility cycle is the basic component of cell migration and consists of a series of well-organized events. These events begin with asymmetric morphology and definition of leading and trailing edges in the cancer cell. Intracellular signals then orient protrusions in the leading edge of the cell, which is followed by a sequence of contraction and detachment movements via integrin-mediated adhesion [45].
Multiple factors can influence cancer cell migration, including the topography of the extracellular matrix (ECM), cell polarity, and cell adhesion [45]. In vitro experiments have demonstrated that tumor cells invading three-dimensional (3D) matrices remodel the ECM microenvironment to migrate. These experiments also have shown that collagen fibers aligned in parallel can promote or at least enable cancer cell invasion, whereas a disorganized, nonlinear matrix reduces invasive behavior. These data suggest that oriented ECMs play a part in directional migration and invasion of cancer cells in vivo [46]. In addition to single-cell migration, collective migration is a principal mode of cell movement in which cells remain connected as they move. Collective migration of cohesive cell groups in vivo is a feature of many invasive tumor types [47].
The invasive phenotype, which distinguishes benign from malignant neoplasms (cancer), is defined by the ability to actively breach or cross tissue barriers, including the bone marrow [48]. This phenotype is manifested at different steps of the metastatic cascade, including escape of cancer cells from the primary tumor, intravasation into and extravasation from the bloodstream, and establishment of a secondary tumor at a distant site. Invasion requires adhesion to and degradation of ECM components and restructuring of the cytoskeleton along with transcriptional and epigenetic changes. Furthermore, invasive cells can adopt different morphogenetic programs, and this transition to a different program is influenced by the tumor microenvironment. When invasive cancer cells utilize a mesenchymal invasion program, the switch from epithelial to mesenchymal cell phenotype is often referred to as epithelial to mesenchymal transition [48]. Recently reported evidence indicated that cells are in cell-cycle arrest when they enter an invasive state. This cell-cycle arrest state is frequently associated with the invasive phenotype acquired via epithelial-to-mesenchymal transition [48–51].
Processing Fresh PCa PDXs for In Vitro Studies
The use of cancer cells derived from PDXs (cPDXs) in laboratory-based research in vitro requires the separation of cancer cells from the mouse stroma. This process constitutes a challenge of differing magnitude depending on the cancer of origin. In the case of PCa, cPDXs do not grow in vitro for long periods, so studies using them must be performed with short-term cultures. cPDXs derived from other malignancies are more amenable to growth through several passages in vitro and may even develop into cell lines. However, during the development of these cell lines, selection of cells for in vitro growth is likely. Thus, short-term cultures are thought to better reflect the phenotype typical of 3D growth and the heterogeneity typical of PDXs than are cell lines. The method we use to isolate cPDXs enriched in PCa cells is described below.
- 1.
Anesthetize PDX-bearing mice by administering isoflurane according to training in the Department of Veterinary Medicine and using an approved inhalation-induction-vapor recovery apparatus. Administer isoflurane at concentrations of 4–5% for induction and 2–3% for maintenance of anesthesia.
- 2.
Kill tumor-bearing host mice via cervical dislocation.
- 3.
Aseptically remove subcutaneous tumors from euthanized host mice as follows: clean the skin over the tumor with 70% ethanol spray, open the skin with a scalpel, and remove the tumor by using scissors to dissect it free from surrounding loose connective tissue (PCa PDXs growing subcutaneously usually do not invade surrounding tissues).
- 4.
Place tumor tissue in a sterile 50-ml conical tube. Rinse the tumor tissue three times using fresh phosphate-buffered saline (PBS) each time.
- 5.
Place the rinsed tumor tissue in a sterile tissue culture dish. Using scissors, remove any remaining connective or other nontumor tissues surrounding the tumor as well as any visible necrotic sections of the tumor.
- 6.
Place the cleaned tumor tissue in a sterile tissue culture dish, and cut it into small pieces with a sterile scalpel. Use a sharp blade to avoid compression damage to the tissue. Place tissue pieces and any spilled cells into a new sterile 50-ml conical tube, wash them with 1× PBS, spin the tissue down in a centrifuge at 300 × g for 5 min, and discard supernatant. This is done under a sterile laminar flow hood.
- 7.
Add Accumax enzyme solution (Innovative Cell Technologies) to pelleted PCa tissue in a conical tube (add enough enzyme to cover the pellet). Incubate the tube for 20 min at 37 °C in a rotation shaker set at 150 rounds/min.
- 8.
Filter out cell clamps from the resulting solution by using a sterile 70-μm-diameter pore cell strainer into a new 50-ml conical tube. This is done under a sterile laminar flow hood.
- 9.
Spin the resulting filtrate in a centrifuge at 300 × g for 5 min at room temperature. Remove and discard the supernatant. Resuspend the pellet in 20 ml of alpha-MEM medium containing 10% fetal bovine serum to neutralize any remaining Accumax enzymes.
- 10.
Rinse tumor solution three to four times, using fresh PBS each time
- 11.
Count the viable cells in the suspension prepared in J using a hemocytometer under a light microscope with a trypan blue assay.
- 12.
Spin the solution prepared in J again in a centrifuge at 300 × g for 5 min at room temperature. Discard the supernatant as described above, and then adjust the tumor cell concentration with growth medium to the desired cell density, and plate it in tissue culture dishes according to the study to be performed.
As previously mentioned, the genetic manipulation (e.g., transfection, silencing of gene expression) of PCa cPDXs is challenging because most of these cPDXs can be propagated in vitro only in short-term cultures. One approach to genetically manipulate cPDXs that is promising is the use of a green fluorescent protein (or green fluorescent protein-like)-tagged virus or bicistronic expression vectors for simultaneous expression of green fluorescent-like proteins and an insert DNA whose expression would lead to modulation of protein expression by overexpression, expression of mutant variants, or gene silencing. GFP tagged CRISPR-Cas9 viral systems can also be utilized for targeted genome editing of cPDXs. Via sequential grafting of infected cPDXs in mice and ex vivo selection of labeled PCa cells using cell sorting, a PDX containing genetically manipulated cancer cells can be established in vivo. This PDX retains the benefits of heterogeneity and in vivo growth, although a certain level of selection of cells probably occurs. This procedure is difficult, and the difficulty is primarily related to the feasibility of growing cPDXs in vitro in monolayers, with PCa cPDXs being the most challenging.
Recent reports described the establishment of organoids from PCa tissue or PDXs. These 3D in vitro procedures are more enabling of genetic manipulation of cPDXs than short-term cultures of cPDXs in monolayers, although these procedures are laborious. Full, detailed protocols for the development of organoids were recently published [52, 53].
Modeling Cell Migration and Invasion In Vitro
Single or collective cell migration in two-dimensional in vitro models can be measured using video microscopy (random motility) or by scraping a monolayer of confluent cells in culture and monitoring their ability to migrate back into the “wounded” area (wound healing assay; directed motility). This method allows for the study of polarization, force generation, and mechanisms of cell-cell cohesion during the movement of confluent monolayers [47, 54, 55].
A Boyden chamber-type system (i.e., a barrier in culture through which cellular invasion can be monitored and quantitated) has been used to model cellular invasion. Several filters through which the cells crawl have been used. More frequently, an 8-μm pore filter is covered with a reconstituted ECM such as Matrigel (Corning Life Sciences and BD Biosciences), which mimics a collagen IV-rich basement membrane, fibronectin, laminin, or a fibrillar collagen I-like meshwork [54]. Similarly, the ability of cancer cells to enter and traverse thick (~2-mm) collagen gels or a monolayer of ECM-producing stromal cells can be used to recapitulate cellular invasion. In each case, chemotactic invasion can be measured by filling the lower chamber of the Boyden chamber-type system with a source of growth factors.
In collective cell migration, several mechanisms polarize the cells as “leader” cells that guide “followers” behind them. The differences between leaders and followers are their clear differences in cell morphology and gene expression. Whereas cells at the leading edge of migration are often less ordered and mesenchyme-like, cells at the rear form more tightly packed assemblies, such as rosettes and tubular networks [47]. Other approaches to studying the invasion of cancer cells include the use of 3D scaffolds overlying ECM alone or with mesenchymal cells. These platforms demonstrate how collective cancer cell invasion is facilitated by both leading cancer cells and leading stromal fibroblasts [56, 57]. For example, authors reported that squamous cell carcinomas that retain epithelial markers cannot remodel the surrounding matrix but instead follow stromal fibroblasts that remodel the ECM [56].
Researchers have developed new technology to monitor cell migration/invasion. For example, the xCELLigence RTCA DP instrument (ACEA Biosciences Inc.) can kinetically measure cell invasion and migration using an electronically integrated Boyden chamber (CIM-Plate 16). Another new development is the 3D spatially organized cancer invasion platform, a microfluidic cancer invasion platform capable of spatially organizing 3D cell-embedded hydrogel matrices while enabling real-time 3D capture of cancer invasion within heterogeneous ECMs [47, 58, 59].
Cancer Cell-Host Cell Interactions
Metastasis requires interaction between cancer cells and their microenvironment. This is particularly striking in the case of PCa, which is the only major malignancy that consistently produces bone-forming metastases, suggesting that factors secreted by PCa cells induce bone formation. In this context, a vicious cycle mediated by soluble factors released by cancer cells and the bone is implicated to support cancer growth. Authors originally described this cycle in the interaction of breast tumor cells with bone cells [60]. Bone homeostasis is maintained by equilibrium of bone formation (mediated by osteoblasts) and bone resorption (mediated by osteoclasts). This balance is disrupted when cancer cells arrive and grow in the bone. Researchers proposed that tumor cells, osteoblasts, osteoclasts, and bone matrices are the four components of the vicious cycle necessary for the initiation and development of bone metastases [60]. Gene expression in tumor cells is modified by factors released from the bone matrix by osteoclast-mediated resorption. In PCa patients, bone lesions are frequently osteoblastic; thus, osteoblast activation is suggested to produce factors that favor the growth of PCa cells [19, 31, 32, 61, 62].
Boyden chamber-type systems have been used to study the effect of soluble factors released by cancer cells and host cell in the cancer cell-host cell interaction. The benefit of these systems is that the effect of soluble factors in the two cell types can be analyzed separately at the end of the study. With the use of this system, we reported that soluble factor release by PCa cells induced the expression of osteoblast-specific factors in osteoblasts and that the Wnt canonical pathway mediated (at least in part) PCa-induced new bone formation (Fig. 7.2) [19, 62, 63].
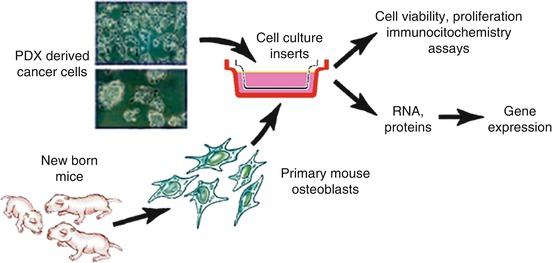
Fig. 7.2
Illustration of a Boyden chamber-type system used to study the interaction between PDX-derived cells (short-term cultures) and primary osteoblasts derived from the calvariae of 4-day-old mice
Another approach to study cancer cell-host cell interactions involves the use of encapsulated cPDX tumor cells in a 3D hyaluronan-based hydrogel. Given the ubiquity of hyaluronic acid in the bone marrow ECM, this approach may be useful in studying PCa cell-bone cell interaction. Use of this approach demonstrated that the hydrogel maintained PDX cell viability with continuous native androgen receptor expression (Fig. 7.3) [26].
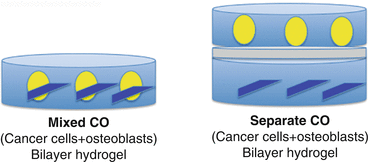
Fig. 7.3
Schematic representation of a 3D hydrogel system for PCa cPDXs and the mouse osteoblast cell line MC 3T3-E1. The hydrogel system comprises the thiolated hyaluronic acid and acrylate-functionalized peptides GRGDS (integrin binding) and PQ (matrix metalloproteinase sensitive). Left, cPDXs are co-encapsulated with osteoblastic (MC 3T3-E1) cells. Thus, cPDXs and MC 3T3-E1 cells are in direct contact. Right, cPDXs and osteoblastic (MC 3T3-E1) cells are encapsulated separately. Thus, their effects are mediated by soluble factors
Tumor Cell Intravasation
The chorioallantoic membrane in chicks is an accessible, blood vessel-rich membrane onto which cPDXs can be placed to test their ability to intravasate into nearby blood vessels and disseminate. This can be studied in vivo in live chick embryos [64, 65] (reviewed in [66]).
Modeling Metastases in Mice In Vivo. Historically, the mouse has been widely used as a model organism to study and molecularly dissect the process of metastasis in vivo. Studies may involve the entire metastatic process when cells are injected orthotopically (in the site of origin of the cancer), and metastatic spread is monitored macroscopically or by labeling the input cells with a transgene-expressing luciferase, which enables them to be tracked in vivo. In general, subcutaneous grafting of PDXs does not result in distant metastasis. Orthotopic engraftment of colorectal carcinoma PDXs has resulted in the development of liver and lung metastases in a proportion of cases [67]. Reports on metastases of breast cancer PDXs implanted orthotopically in the mammary fat pad in mice included different metastatic outcomes. In one report, metastases occurred at a very low rate (3 of 144 mice) after long periods of observation following resection of primary tumors [68]. Furthermore, these metastases did not result in a more aggressive metastatic phenotype upon retransplantation into new host tumors [68]. In two other reported studies, lymph node and lung metastases were frequently detected [69], with lung metastases found in 48% of cases [70]. Recently, authors reported that thin tissue slice grafts of renal cell carcinomas had the potential to metastasize to clinically relevant sites, including the liver, lung, and bone. Also, lymph node metastases were reported to develop after intramuscular implantation (in the quadriceps of mice) of PDXs derived from highly aggressive squamous cell carcinomas of the uterine cervix [71].
Frequently, researchers design their studies to focus on specific aspects of the metastatic process in vivo. For example, cell motility and invasion in vivo can be studied using intravital microscopy. The ability of cells to reach specific organs and grow can be studied with intracardiac injection of cancer cells into the left ventricle (termed “experimental” metastasis). In this scenario, the initial steps of metastasis are bypassed. Finally, the interaction of cancer cells with the host cells at the metastatic site can be studied using direct injection of the cancer cells into the organ subject of study. This strategy does not provide information about the metastatic process but does provide important information on the tumor/stroma interactions that lead to tumor growth at the metastatic site.
These experimental procedures have strengths and shortcomings, and determining the value of each of these methods comes down to the scientific question that is being addressed and how well the methods for answering the question were selected. For example, intracardiac injection of cancer cells into the left ventricle will provide important evidence regarding the preferred site of metastasis of a given cPDX and how this tropism can be altered by genetically manipulating the injected cells. However, the effects of genetic manipulation of the injected cells in the initial steps of metastasis cannot be studied using intracardiac injection. Also, the effects of genetic manipulation of the injected cPDX on cancer cell-host cell interaction at the metastatic site cannot be accurately assessed because the pattern of metastasis is unpredictable. A more informative method for the latter would be direct injection of a cPDX into the metastatic site because the subject of the study is controlled. For preclinical/co-clinical studies, direct injection of a cPDX into the organ of interest is preferred if the purpose of the study is to identify means of controlling the growth of established metastases. If the goal is to prevent the development of metastases, either orthotopic or intracardiac injection would be adequate. These methods are described below.
Intravital Microscopy
An approach to studying cell motility and invasion in vivo is labeling injected cells with a vital dye or a green fluorescent protein–tagged transgene, which allows for evaluation of the initial steps of metastasis using intravital microscopy. However, this approach can only be used when the tissue of interest can be accessed directly through an imaging window or surgically [72–74].
Briefly, multiphoton microscopy is based on the use of two or more low energy photons rather than a single higher-energy photon in examination of a sample. As a result, the focus is only on a dot, and no excitation or bleaching occurs above or below it. A significant advantage of using lower-energy excitation is penetration. Both deep-tissue imaging [75, 76] and imaging resolution at the single-cell level for monitoring of tumor cell behavior during metastasis [77–79] are possible thanks to multiphoton excitation. This technology allows for imaging even at the subcellular level, resulting in definition of parameters such as cell kinetics, morphology, the presence and nature of protrusions, and the proliferative state to further understand tumor heterogeneity and better outline its role in the different steps of metastasis.
A great benefit of assessing tissues using this technology is that they generate intrinsic signals on their own. For example, collagen fibers making up the ECM in bone tissue become optically accessible by second harmonic generation. Additionally, third harmonic generation permits visualization of cell and tissue interfaces, such as water-lipid interfaces, including adipocytes, microvesicles, exosomes, and nerves, owing to the high density of myelin in them. Technologies that do not require external probes include optical frequency domain imaging for deep, continuous imaging over time [80] and Coherent anti-Stokes Raman spectroscopy for studying lipid distribution [81]. Even moving nonfluorescent host cells can be imaged as a “side effect” of fluorescent tumor cells caused by light scattering by the resident cells, particularly immune cells, that form shadows outlining their shapes [82]. Many of the components of the tumor microenvironment are readily capable of being imaged.
The role of the microenvironment in metastasis can be studied via simultaneous imaging (using intravital microscopy) of stroma and tumor cell components stained with dyes in multiple colors [77, 83]. A wide variety of vital dyes can be injected directly into mice, enabling staining of specific cell types and/or subcellular structures endogenously [84]. For example, AngioSense (PerkinElmer), fluorescent dextrans, quantum dots (nanocrystals), and fluorescently labeled lectins that selectively bind to endothelial cells [85] can be used to visualize blood vessels and lymph nodes. Also, distinct cell populations, such as macrophages and monocytes, can be labeled with iron oxide nanoparticles. Furthermore, the activity of some enzymes can be examined, such as metalloproteinases using MMPSense (PerkinElmer) and cathepsins using ProSense®. Other agents, such as blue Hoechst 33342, are used for ex vivo cell staining. Stable expression of the fluorescence ubiquitination cell cycle sensor reporter system can be used to monitor the proliferative status of tumors implanted subcutaneously in mice [86]. OsteoSense 680 EX (PerkinElmer) is an in vivo imaging agent used to study bone growth and resorption by defining areas of microcalcification and bone remodeling using fluorescent bisphosphonate. Photoswitchable flourophores such as Dendra2, PSmOrange, and Kaede, which act by switching the color of a subset of cells, are applied to tracking the position and motility of tumor cells in subsequent imaging over multiple days. With the use of this feature, studies of primary cancer cell colonization at distant organs in a mouse orthotopic mammary cancer model revealed that some tumor microenvironments could stimulate metastatic behavior, whereas others exhibited lower rates of tumor cell invasion and intravasation [87].
Single-cell resolution studies identified different modes of migration of tumor cells—mesenchymal, ameboid and blebby, streaming, and collective—which can be found together in strands, clusters, or single cells in the same lesion. Multiphoton excitation allows for optical examination of morphologic cell changes. Reduction in compactness of ECM network during carcinogenic progression frees up cell movement and morphology. In general, the shapes of the tumor cells are not elongated, rigid, or fibroblastic anymore after reduction of ECM network but tend to be ameboidal with extending lamellipodia at their edges [76, 79]. At the subcellular level, the arrangement of the cytoskeleton components in migrating cells, assayed via fluorescent labeling of the myosin light chain in the actomyosin, demonstrated how the myosin light chain localizes to extending protrusions in motile cells and that these protrusions, or invadopodia, cause ECM deformation [88]. Signaling pathways involved in migration in situ also can be visualized.
As described previously, intravital microscopy can be used only when the tissue of interest can be accessed directly through an imaging window [72, 74]. Researchers have developed different approaches to accessing tumor tissue optically. The dorsal skinfold chamber is one widely used technique, although a disadvantage of this method is that the tumor grows ectopically (except for skin-derived cancers) and the size of the tumor is limited to that of the slide inside the chamber. Also, investigators developed a cranial window implant to image the brain cortex, which is facilitated by the shallow thickness and thus the relative transparency of the brain surface [89]. Abdominal imaging windows are used to study tumors in the intestine, pancreas, and liver. Mammary imaging windows are used to recapitulate breast tumors orthotopically. Other organs can be made more accessible using surgical procedures with variable degrees of invasiveness. A small skin incision enables imaging of popliteal lymph nodes [90], whereas the liver can be visualized via previous exteriorization and superfusion with physiological saline solution [91]. In developing models of other organs, such as the kidney, pancreas, spleen, and heart, researchers must consider inflammatory responses as well as muscle contractions, respiration, and blood vessel pulses, which can cause unwanted movements during imaging that must be restrained under deep anesthesia and with some form of physical immobilization.
Further methods to accessing tumor tissue optically and model the specific microenvironmental site in an ectopical imaging chamber context have been introduced. The application of engineered scaffolds to mimicking the bone as a preferred site of metastasis of breast and PCa and as a key site of stem cell development has been useful. These are called implantable microenvironments and can be visualized in a dual skinfold chamber [92]. Tissue engineering is another field that will help further model metastases for the study of invasion, migration, and cancer cell-host cell interaction.
Intracardiac Injection of Tumor Cells into the Left Ventricle
Intracardiac injection of tumor cells measures experimental metastases and is aimed at the direct release of cPDXs into the left ventricle so that they can reach all organs via the blood vessels. The cPDXs can be labeled with luciferase so that they can be visualized when they grow in distant organs. The methods we use to implant tumor cells into the left ventricle are described below:
- 1.
Using the solution prepared in J (processing fresh PCa PDXs for in vitro studies), spin it again in a centrifuge at 300 × g for 5 min at room temperature. Discard the supernatant, and then adjust the tumor cell concentration by adding 1× PBS as needed to produce a final suspension containing 0.5 × 106 to 1.5 × 106 tumor cells per 50–100 μl. Place the tube containing the final suspension on ice in preparation for implantation of the cells into mice as described below.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree