Gene
Smoking status
Histology
Genetic alteration
ROS1
Primarily never smokers
AD
ROS1 translocations
RET
Primarily never smokers
AD
RET translocations
HER2
Primarily never smokers
AD
HER2 mutations
HER2 amplification
BRAF
Primarily smokers
AD/SCC
BRAF mutations
cMET
N/A
AD/SCC
cMET overexpression,
amplification, and mutation
PI3K pathway
More in smokers
SCC/AD
PTEN loss
PI3K amplification
PI3K mutations
AKT mutations
DDR2
–
SCC
DDR2 mutation
FGFR1
Primarily smokers
SCC
FGFR1 amplification
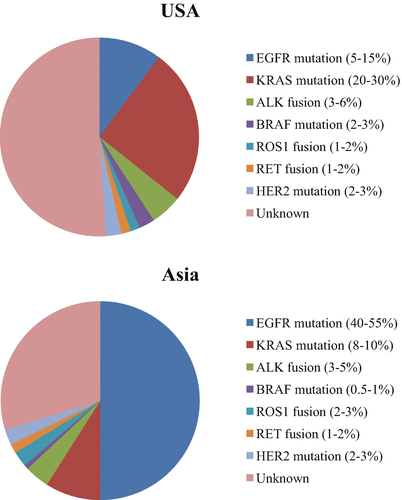
Fig. 12.1
The frequencies of oncogenic driver mutations in LADC (Data from patients in the USA and Asia were taken by previous reports (13–14). LADC lung adenocarcinoma)
Kris et al. have reported a median survival of 3.5 years (interquartile range [IQR], 1.96–7.70) for 260 patients with an oncogenic driver and genotype-directed therapy compared with 2.4 years (IQR, 0.88–6.20) for 318 patients with any oncogenic drivers who did not receive genotype-directed therapy [15]. This showed that those individuals with drivers receiving a matched targeted agent lived longer and that randomized trials are required to determine whether targeting therapy based on oncogenic drivers improves survival. Therefore, it will be necessary to develop systematic genomic testing algorithms to identify defined subsets of patients with NSCLC for whom effective drug therapies are available either commercially or through clinical trials.
In this article, we focus on these minor-driver mutations in NSCLC, regarding their characteristics, frequencies, mechanism whereby agents target them, and clinical evidence relating to the use of these agents.
12.2 Minor Mutations
12.2.1 RET
The RET receptor tyrosine kinase is encoded by the RET proto-oncogene localized on human chromosome 10q11.2. It is believed to be required for development of the kidneys and enteric system, as well as for the differentiation and survival of neurons [17, 18]. RET is the signaling receptor for the glial cell-derived neurotrophic factor (GDNF) family of ligands: GDNF, neurturin (NRTN), persephin (PSPN), and artemin (ARTN) [18]. After ligand binding, the intracellular kinase domain is activated, followed by autophosphorylation of intracellular tyrosine residues. These phosphotyrosine residues then serve as a platform to which downstream signaling proteins carrying SRC homology 2 (SH2) or phosphotyrosine-binding (PTB) domains bind and transmit signals into the cell, leading to the activation of RAS/ERK1/2 and PI3K/AKT pathways [19].
RET fusion-positive cases have been found in about 1–2 % of LADC patients in Asian and European populations [5–7, 20]. All reported lung tumors with RET rearrangements have exhibited LADC histology. Where overlap was evaluated, RET rearrangements have been shown to occur in tumors without other common oncogenic drivers such as EGFR, ALK, and ROS1. The most commonly reported fusion gene was KIF5B-RET [6]. Other commonly observed fusion genes are CCDC6-RET, NCOA4-RET, and TRIM33-RET.
RET rearrangements may be identified by fluorescence in situ hybridization (FISH), reverse transcription-polymerase chain reaction (RT-PCR), or next-generation sequencing. The use of immunohistochemistry (IHC) for RET visualization has had variable results and is not popular for identification of RET fusion genes [21].
RET rearrangements result in the formation of fusion products, which are capable of undergoing constitutive dimerization, leading to subsequent ligand-independent kinase activation, potentially resulting in neoplastic transformation. RET rearrangements are oncogenic in vitro and in vivo. Preclinical studies have demonstrated that lung cancer cell lines with RET fusions may be sensitive to multi-kinase inhibitors such as vandetanib, sunitinib, and sorafenib [5, 20]. Other preclinical evidences have demonstrated that cell lines with RET fusion genes had transforming capacity and enhanced sensitivity to vandetanib and other RET inhibitors [22, 23].
A variety of agents targeted against RET pathways have been studied in other malignancies. These include vandetanib, cabozantinib, sunitinib, sorafenib, fostamatinib, and ponatinib which have shown good responses, especially in RET rearrangement tumors [19]. However, studies with RET inhibitors in NSCLC are lacking, because no inhibitor specific for RET is available. There are case reports of vandetanib leading to remission 4 weeks after starting therapy in LADC; KIF5B-RET positive fusions following chemotherapy, surgery, and radiation [24]; and partial response (PR) for 4 months in CCDC6-RET fusion-positive LADC [25]. A prospective phase II trial studying the role of the multi-kinase inhibitor cabozantinib in three RET fusion-positive NSCLC patients showed PR to treatment, and the third patient had a stable disease (SD). All three patients were progression-free at the time of reporting (4–8 months) [7]. It has been reported that the use of sunitinib in a case of KIF5B-RET fusion-positive NSCLC provided clinically significant disease control for 10 weeks until sunitinib was discontinued [26]. To date, five independent, open-label, single-arm, phase II studies have been conducted to evaluate the efficacy of sunitinib (NCT01829217), cabozantinib (NCT01639508), ponatinib (NCT01813734), vandetanib (NCT01823068), and lenvatinib (NCT01877083) in NSCLC with RET rearrangements (Table 12.2). In Japan, the Lung Cancer with RET Rearrangement (LURET) study (UMIN000010095) has evaluated the efficacy of vandetanib in 19 patients with RET fusion gene-positive NSCLC (Table 12.2). Among 17 eligible patients, nine (53 % [95 % CI 28–77]) achieved an objective response, with a median progression-free survival of 4.7 months (95 % CI 2.8–8.5) [27]. Similar impressive clinical activity has been observed in phase II study of carbozantinib in RET-positive NSCLC. The overall response was 28 % (95 % CI 12–49), which met the primary endpoint, and the median progression-free survival was 5.5 months (95 % CI 3.8–8.4) [28].
Table 12.2
Phase II clinical trials of RET-targeting therapies with RET fusion gene-positive NSCLC
Trial number | Treatment | Location | Study design | Primary end point | Enrollment |
|---|---|---|---|---|---|
NCT01639508 | Cabozantinib | USA | Open-label, single arm | Response rate | 26 |
UMIN000010095 | Vandetanib | Japan | Open-label, single arm | Response rate | 19 |
NCT01823068 | Vandetanib | Korea | Open-label, single arm | Response rate | 18 |
NCT01877083 | Lenvatinib | Global | Open-label, single arm | Response rate | 20 or more |
NCT01813734 | Ponatinib | USA | Open-label, single arm | Response rate | 20 |
12.2.2 ROS1
ROS1 is a human receptor tyrosine kinase, encoded by the ROS1 gene which is closely related to the ALK gene [29]. ROS1 has considerable amino acid homology with ALK [30]. Oncogenic activation of ROS1 as a result of different chromosomal rearrangements has been identified in a variety of human tumors including LADC, where it has been reported in approximately 5 % of such cases [11, 31]. ROS1 rearrangements tend to occur in LADC with solid, papillary, cribriform, or signet ring cell histologic patterns, tend to produce mucin, and tend to arise in patients who are younger and never smokers [11]. ALK and ROS1 fusions are nonoverlapping with other known drivers in lung cancer, such as KRAS and EGFR mutations [11]. Several fusion genes have been identified including CD74-ROS1, SLC34A2-ROS1, EZR-ROS1, TPM3-ROS1, and SDC4-ROS1; the ROS1 kinase gene is retained in all of these fusion events, and the expressed fusion genes are believed to play a role in carcinogenesis [11, 31, 32]. The mechanisms of oncogenic transformation provoked by these fusion genes are believed to involve upregulation of the phosphatase SHP-2, the PI3K/AKT/mTOR pathway, the JAK/STAT pathway, and the MAPK/ERK pathway [31].
The presence of ROS1 rearrangements may be detected by FISH with the ROS1 break-apart probe and by RT-PCR [5, 7, 21]. FISH testing is not dependent on the specific fusion partner. Specific fusion partners are detected by RT-PCR [11]. IHC can be used to screen for positive ROS1 which can then be confirmed by FISH. IHC has been reported to be highly sensitive for ROS1-positive LADC confirmed by FISH and RT-PCR with strong diffuse expression; however, false-positive IHC has been reported to occur in some ROS1-negative LADC with results confirmed by FISH as required [33].
Preclinical studies have reported that ROS1 fusions are associated with sensitivity to tyrosine kinase inhibitors that have off-target activity against crizotinib [11]. Clinically, two patients with tumors harboring ROS1 fusions have shown partial responses to crizotinib [11, 31]. A retrospective analysis of 1073 NSCLC cases demonstrated no difference in OS between ROS1 rearrangement positive and negative subgroups, but suggested sensitivity of ROS1 rearrangement positive cell lines [11].
A phase I study of the efficacy of crizotinib in ROS1-rearranged NSCLC showed marked antitumor activity, with an objective response rate of 72 % (95 % CI, 58–84) and median progression-free survival (PFS) of 19.2 months (95 % CI, 14.4 to not reached). Although several types of ROS1 rearrangement have been described, this study did not find any correlation between the type of rearrangement and response to therapy [34]. In a retrospective European case study, 31 ROS1-positive NSCLC cases treated with crizotinib were retrospectively reviewed, and an objective response rate of 80 % and 9.1-month median PFS were calculated in this cohort [35].
Acquired resistance to crizotinib has been observed in LADC with ROS1 fusion genes, as with ALK-positive LADC. The mechanism of acquired resistance to ROS1 inhibition has been reported to involve an acquired mutation in CD74-ROS1 fusion, whereby a glycine-to-arginine substitution occurs at codon 2032 in the ROS1 kinase domain [36]; in addition, a different mechanism may involve activation of alternative signaling via the EGFR pathway [37]. Furthermore, preclinical studies have reported that foretinib is a potent ROS1 inhibitor [38].
12.2.3 BRAF
The oncogene BRAF encodes a serine/threonine kinase that lies downstream of RAS protein in the RAS-RAF-MEK-ERK signaling pathway [39]. BRAF mutations are seen in about 50 % of melanomas, where BRAF V600E is a driver mutation that can be effectively targeted with selective BRAF and/or MEK inhibitors [40]. BRAF mutations have been found in about 1–5 % of patients with NSCLC [41–43]. In contrast to melanoma, the BRAF mutations found in NSCLC were V600E (50 %), G469A (39 %), and D594G (11 %) [42]. V600E BRAF mutations occurred more frequently in women and never smokers, whereas non-V600E mutations occurred more commonly in current and former smokers. The BRAF V600E genotype has been associated with more aggressive tumor histology and poorer prognosis compared to non-V600E genotypes [44].
BRAF V600E mutations can be detected with targeted next-generation sequencing [45]. IHC using the VE1 antibody has also been reported as a successful screening tool for BRAF V600E mutation in LADC [46].
Many of these non-V600E mutations show only intermediate or low kinase activity, and preclinical studies have suggested that non-V600E BRAF-mutated kinases were resistant to BRAF-targeted therapy, although some may be sensitive to downstream pathway inhibitors such as MEK inhibitors [47]. In a histology-independent, biomarker-selected, early phase II basket study of vemurafenib, this agent showed modest antitumor activity in cancers that sporadically express BRAF V600 mutations [48], whereas the BRAF G469L mutant LADC did not respond to vemurafenib [49]. These studies suggested that BFAF V600E mutations were predictive biomarkers for therapy of LADC with vemurafenib. The selective BRAF V600E mutant kinase inhibitor dabrafenib has increased PFS in BRAF V600E-mutated metastatic melanoma compared to conventional therapy [50]. Two patients with BRAF V600 mutant NSCLC are reported to have had a PR to dabrafenib [51]. Currently, agents targeting BRAF or downstream effectors in ongoing clinical trials include dabrafenib for patients with NSCLC and prospectively identified BRAF V600E mutations: the MEK inhibitor, trametinib, for patients with non-V600E BRAF mutations; and dasatinib for patients with NSCLC and inactivating or uncharacterized BRAF mutations (NCT01336634, NCT01362296, and NCT01514864, respectively).
Multiple mechanisms of acquired resistance to targeted BRAF V600 inhibitors have been described in melanoma, including upregulation of receptor tyrosine kinases, activation of the AKT pathway, and acquired mutation in NRAS [52]. Recent data suggest that inhibition of BRAF V600E can activate feedback leading to increased activity of and dependence on RAS [53]. In a case of LADC, it was reported that the acquired KRAS G12D mutation was primarily responsible for acquired dabrafenib resistance in this patient [54].
12.2.4 HER2
HER2 mutations have been reported in approximately 2 % of NSCLC patients, with these generally lacking other EGFR, ALK, and KRAS mutations. HER2 mutations are more prevalent in LADC from patients who are never smokers and are more common in Asians and females. Similarly, HER2 gene amplification has been found in approximately 2 % of NSCLC patients identified by FISH using criteria for HER2 amplification in breast cancer [55, 56].
HER2 mutations mostly occur in exon 20 as in-frame insertions, leading to constitutive activation of the receptor and downstream AKT and MEK pathways [57, 58]. HER2 mutations respond to the genetic driver definition, and preclinical models have proved the concept of the transforming property of such a genetic alteration [59]. HER2 mutations may be more relevant in lung carcinogenesis than HER2 amplification or overexpression. Some ongoing clinical trials are enrolling HER2-mutated NSCLC patients, mixed together with HER2-amplified or EGFR-mutated NSCLC patients. Clinical trials with HER2 mutants in NSCLC have shown promising preliminary results for therapy with afatinib [55, 60], trastuzumab [55], dacomitinib [61], and neratinib plus temsirolimus [62].
12.2.5 NTRK1
Chromosomal rearrangements involving neurotrophic tyrosine kinase 1 (NTRK1) occur in a subset of NSCLC, leading to expression of an oncogenic tropomyosin-related kinase (TrkA) fusion protein. NTRK1 fusion genes in NSCLC were first described among a population of LADC patients in the absence of detectable EGFR or KRAS mutation, or ALK or ROS1 fusion genes [63]. Two different NTRK1 fusions have been described in NSCLC using next-generation sequencing, namely, MPRIP-NTRK1 and CD74-NTRK1 [63]. Preclinical evidence supports the role of these fusions in TrkA autophosphorylation leading to oncogenic processes [63]. It was indicated that entrectinib, an orally available small molecule inhibitor of TrkA, TrkB, TrkC, ROS1, and ALK, showed significant antitumor activity in a patient with NSCLC harboring an SQSTM1-NTRK1 gene rearrangement [64].
12.2.6 NRG1
The CD74-NRG1 fusion genes which are a chimeric transcript fusing the first six exons of CD74 to the exons encoding the EGF-like domain of the neuregulin-1 (NRG1) III-β3 isoform have been shown to occur specifically in invasive mucinous adenocarcinomas (IMAs) of never smokers, a tumor type that is otherwise associated with KRAS mutations [65]. It has been reported that NRG1 fusions were present in approximately 1.7 % of lung adenocarcinomas and in 17.6 % of KRAS-negative IMAs [65, 66]. The CD74-NRG1 fusion activated HER2/HER3 signaling, whereas the EZR-ERBB4 and TRIM24-BRAF fusions constitutively activated the ERBB4 and BRAF kinases, respectively.
12.2.7 FGFR1/FGFR3
Lung SCCs have fewer treatment options than those with non-SCC NSCLC. Thus, driver mutations are emerging that may be linked to outcomes with targeted therapies in SCC. Altered genes include FGFR1 and DDR2 as well as PIK3CA. In addition, results from a recent large-scale genomic study in lung SCC have added a variety of potential therapeutic targets that await validation in prospective clinical trials.
The fibroblast growth factor receptor (FGFR) 1/FGFR3 genes encode one member of the FGFR TK family, which includes four kinases: FGFR1, FGFR2, FGFR3, and FGFR4. FGFR TKs belong to the immunoglobulin superfamily and act as receptors for the various fibroblast growth factors (FGFs). Amplification or activation of FGFR1 has been reported in lung SCC from former/current smokers [67, 68]. The Cancer Genome Atlas (TCGA) reports demonstrated FGFR3_missense mutations (3 %), amplifications (0.6 %), fusions (2.2 %), and deletions (1.7 %) in lung SCC and FGFR3 amplifications (1.3 %) and a single mutation event to S779R (0.4 %) in LADC [9, 69]. FGFR3–TACC3 fusions were identified in 0.5 % of cases in lung adenocarcinoma [70, 71]. In other reports, lung SCCs were most notable for their 9 % frequency of FGFR1 amplification, which is in contrast to only 4 % of LADC harboring any FGFR abnormality [72]. Preclinical studies have shown that cancers harboring FGFR3–TACC3 fusions and other FGFR3 fusions are sensitive to pan-FGFR inhibitors and FGFR-selective agents [8, 70]. In addition, such studies indicated that cancer cells with amplified FGFR1 could display addiction to FGFR signaling [69]. Clinical trials with FGFR inhibitors are currently under way.
Related posts:
 Small Cell Lung Cancer and Molecular Targeted Therapy
Management of Adverse Effects by Molecular Targeted Therapy and Immunotherapy
Small Cell Lung Cancer and Molecular Targeted Therapy
Management of Adverse Effects by Molecular Targeted Therapy and Immunotherapy
 Screening Lung Cancer with Low-Dose CT Combined with Molecular Markers
Screening Lung Cancer with Low-Dose CT Combined with Molecular Markers
 Health-Related Quality of Life in Molecular Targeted Therapy
Health-Related Quality of Life in Molecular Targeted Therapy
 EGFR Mutant
EGFR Mutant
 PET-CT, Bio-imaging for Predicting Prognosis and Response to Chemotherapy in Patients with Lung Cancer
PET-CT, Bio-imaging for Predicting Prognosis and Response to Chemotherapy in Patients with Lung Cancer
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


