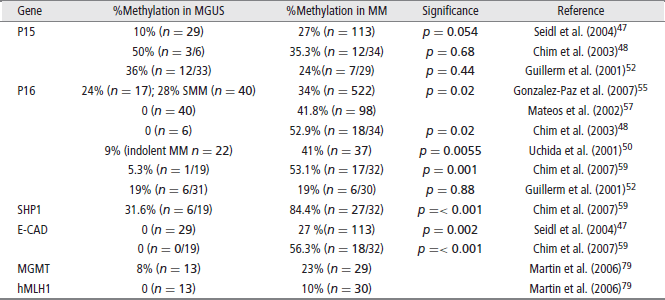
TABLE 2 | Methylation implicated in malignant transformation of monoclonal gammopathy of uncertain significance (MGUS) to Multiple Myeloma (MM)
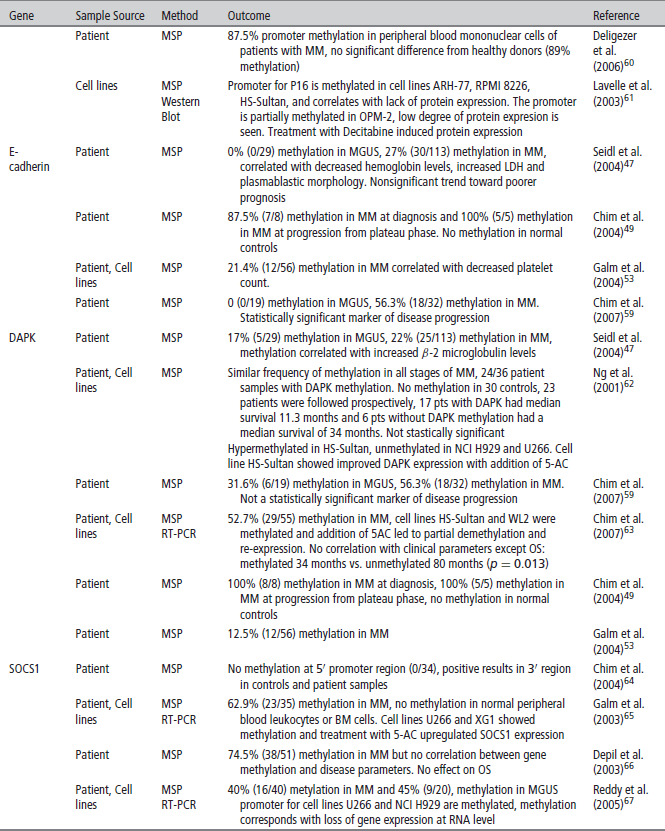
P16 (CDKN2A)
P16 is an inhibitor of cyclin-dependent kinases (CDKs) and inhibits CDKs 4 and 6. P16 promoter methylation leads to gene silencing that may cause cell cycle activation through upregulation of CDKs 4 and 6, resultant plasma cell proliferation and disease progression. P16 is found to be methylated in 19–53% of the genomes of patients with MM.52,59 P16 inactivation is most often achieved through methylation in MM, as P16 deletions and mutations are rarely found. Although some studies demonstrate conflicting results, P16 is very likely associated with the pathogenesis of MM. Furthermore, it has been shown that patients with a methylated P16 gene had three times the amount of plasma cells in S-phase than patients that did not have a methylated P16 gene, demonstrating its tumor suppressor functions.57 Given the finding of significant increased CpG island methylation in MM as compared to MGUS, silencing of P16 is also implicated in the progression of MGUS to MM (Table 2). Multiple studies have found a correlation with a silenced P16 gene and overall worse prognosis.53,55 In addition, P16 has been correlated with elevated beta 2 microglobulin serum levels, and high C-reactive protein values, both markers of bad prognosis for overall survival (OS) in myeloma.
E-Cadherin
E-Cadherin (E-CAD), another tumor suppressor gene commonly studied in MM, is a gene that expresses a cell adhesion molecule. When methylated, downregulation of E-CAD could theoretically decrease cell adhesion within tissues allowing for more cellular motility and potential metastasis. E-CAD is methylated in 27–56% of MM patients. It also appears to play an important role in the progression from MGUS to MM. Two studies that looked at E-CAD found no methylation in patients with MGUS but found a high percentage of methylation (27% and 56%) in patients with MM.47,59 In addition to being a marker of disease progression, E-CAD has been associated with poor prognostic markers such as a decreased hemoglobin (p = 0.024) increased lactate dehydrogenase (LDH)(p = 0.035) and a lower platelet count. It was also found to be correlated with a plasmablastic morphology (p = 0.012) of myeloma cells, representative of a markedly metastatic form of the disease.47,53
SHP and SOCS
The Jak/STAT pathway is a cellular signaling pathway stimulated by interleukin-6 (IL-6), a major growth factor for survival of myeloma cells. This pathway can be negatively regulated by three families of proteins. Two of these, the SH-2 containing phosphatases (SHPs) and the suppressors of cytokine signaling (SOCS), have been found to be methylated and silenced in patients with MM. Decreased expression of these negative regulators leads to overactivation of JAK-STAT signaling that can lead to increased survival and proliferation signals. Methylation frequencies of SHP vary, ranging from < 20% to 79.4%. SHP has also been correlated with disease progression.64,67 SOCS-1 is methylated with frequencies ranging from 0 to 62.9% and appears to be a very early event in MM and has not been shown to be a marker of disease progression.
Death-Associated Protein Kinase (DAPK)
DAPK is a proapoptotic calcium/calmodulin-regulated serine/threonine kinase that has been seen to be methylated in 12.5–67% of MM samples.53,62 DAPK hypermethylation is seen in similar frequency in both MM and MGUS indicating that it may be an early event, similar to SOCS1, in MM pathogenesis.59,62 Patients with DAPK hypermethylation have demonstrated a trend toward a poorer response to treatment and a statistically significant poorer OS, p = 0.013.80 In addition, methylation of DAPK was correlated with increased B2 microglobulin (p = 0.015) levels and lower platelet counts (p = 0.03).53
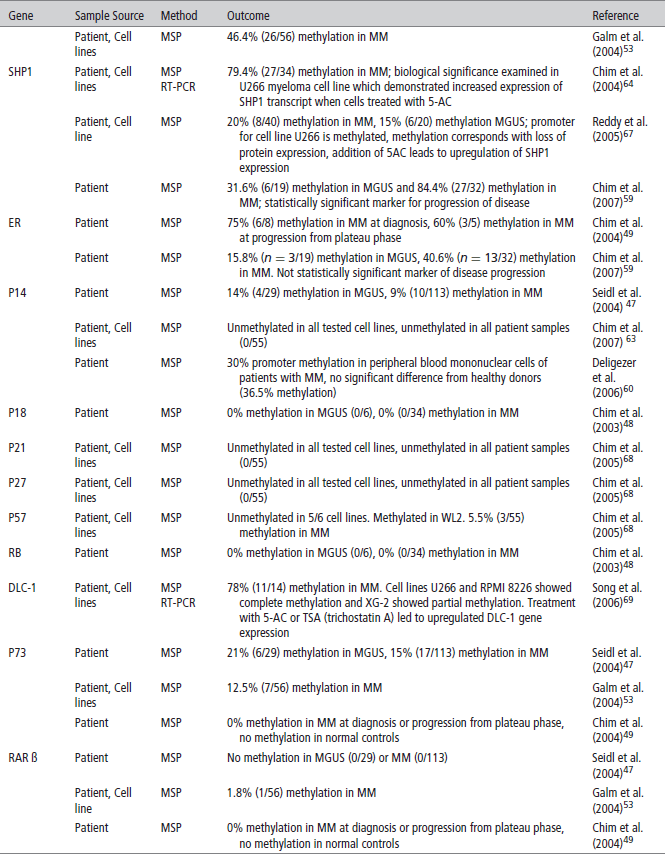
DNA Damage Repair Genes
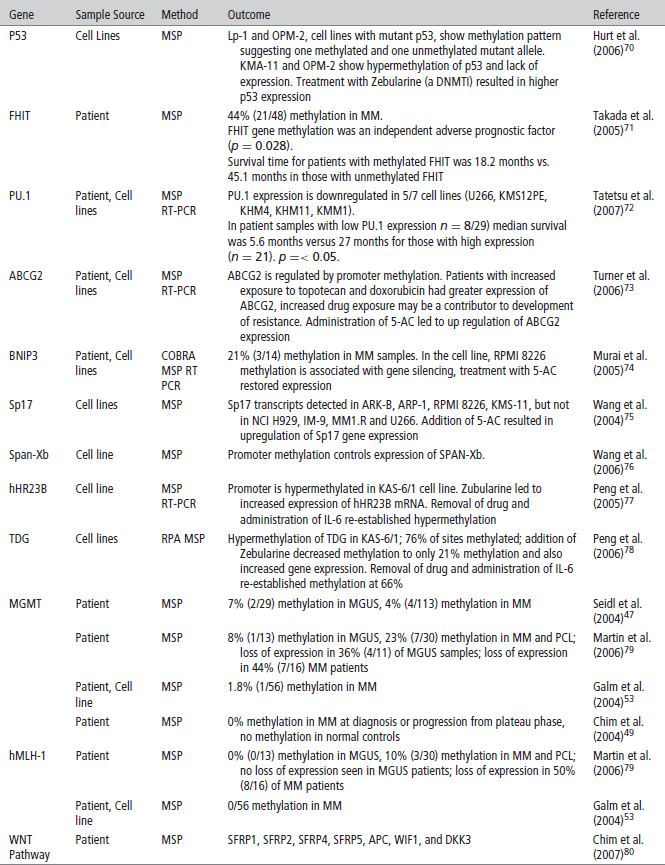
The silencing of DNA damage repair genes may play an important role in the pathogenesis of MM by leading to genomic instability and accumulation of genetic mutations. hHR23B, a nucleotide excision repair protein that removes helix distorting damage, was shown to be methylated and silenced in myeloma-derived KAS-6/1 cells. Furthermore, when Zebularine, a potent demethylating agent, was added, an increase in hH23B mRNA expression was observed.77 TDG, thymine-DNA glycosylase, a base excision repair protein, has also shown to be hypermethylated in myeloma cell lines.78 O6-methylguanine DNA methyltransferase (MGMT) and human mutL homolog 1 (hMLH1), both important in DNA repair, have been shown to be methylated with increased frequency in MM patients when compared to MGUS (Table 2).79 This may be an important link in our understanding of myeloma pathogenesis but more clinical data on the prevalence of DNA damage repair genes in MM is needed.
In contrast, fewer genes have been found to be hypomethylated in myeloma. JAG2 is a protein that promotes the survival and proliferation of hematopoetic progenitors and has been shown to be hypomethylated and overexpressed in myeloma. JAG2 has been shown to induce increased expression of IL-6, a major growth factor that promotes plasma cell proliferation in MM.83
Etiology of Aberrant Methylation in Myeloma
The etiology of aberrant methylation is still unclear, though studies have linked both hyper- and hypomethylation to altered DNA methyltransferase activity. Patterns of DNA methylation are determined during embryogenesis by three active DNMTs: DNMT1, DNMT3a, and DNMT3b. DNMT1 works mostly on hemimethylated DNA and is important in maintaining DNA methylation during replication.84 DNMT3a and 3b are responsible for de novo methylation seen in early embryos, and thus play an important role in development as well as disease states.85 It has been shown that DNMTs are both over and underexpressed in various malignancies. DNMT1 has been found to be overexpressed in MM. It has been shown that IL-6 induces activation of STAT3, initiating transcription of transcription factor FLI-1 (friend leukemia integration-site 1) that in turn increases expression of DNMT1 in KAS 6/1cells. An increase in IL-6 in myeloma, correlating with an increase in DNMT1, may contribute to the etiology of hypermethylation found in myeloma.86
Implications for Treatment
Because various studies have shown multiple genes to be hypermethylated in MM, inhibitors of DNA methyltransferase (DNMTIs) are potential new therapeutic agents for this disease. Most data for the therapeutic efficacy of these agents are derived from in vitro studies involving cell lines that examine one or two methylated genes and demonstrate that addition of DNMT inhibitors in vitro cause demethylation and increased gene transcription of the gene of interest. This has been shown for SOCS1, DAPK, SHP1, and multiple other genes (see Table 1). In addition, it has been shown in vitro that doxorubicin and bortezomib synergistically enhance 5-azacytidine induced MM cell death, suggesting that DNA methylation may be involved in resistance to conventional chemotherapeutic agents as well.87 It should be noted, that to date none of the available DNA demethylating agents are able to target aberrant methylation of specific genes. Clinical trials with the DNMT inhibitors are needed to understand its possible use in MM and are currently in process.
As mentioned above, histone deacetylation is a second important epigenetic factor that contributes to gene silencing along with DNA hypermethylation at CpG islands in promoter regions. It is believed to help maintain heterochromatin tightly wound to prevent its transcription. When 5-Aza-2′-Deoxycytidine (DNMTI) and Trichostatin A, a histone deacetylase inhibitor (HDI) were used together to treat cell lines, gene expression studies revealed additional cancer related genes that were induced when compared to either treatment alone. These genes include CDKN1A/p21, WIG1, BIK, CGREF1, JUP, and IGFBP3. This demonstrates that these two drugs may work synergistically to reverse epigenetic silencing.88 There are currently multiple trials in process looking at HDIs with first-line MM drugs such as lenalidomide and bortezomib, but future trials using both DNMTIs and HDIs are needed to assess the potential synergistic action of this therapy.
GENOME-WIDE ANALYSIS OF DNA METHYLATION CAN UNCOVER NEWER EPIGENETIC TARGETS IN CANCER
Most of the studies on aberrant epigenetic marks in myeloma and other malignancies have been single locus studies. These studies have also focused on examination of CpG islands of genes selected on the basis of previous studies or investigator preference. In studies performed in myelodysplasia, a hematologic malignancy that responds to treatment with azacytidine, changes in the methylation status of selected genes post-treatment have not correlated with clinical responses.89 Thus, a genome-wide, unbiased examination of DNA methylation is more likely to reveal changes that may have pathologic as well as prognostic implications. The recent advent of genome-wide assays for cytosine methylation analysis does indeed show widespread epigenetic dysregulation in cancers.90,91 It is critical that these unbiased approaches are used in future studies to uncover genes that are important in pathogenesis of cancer.
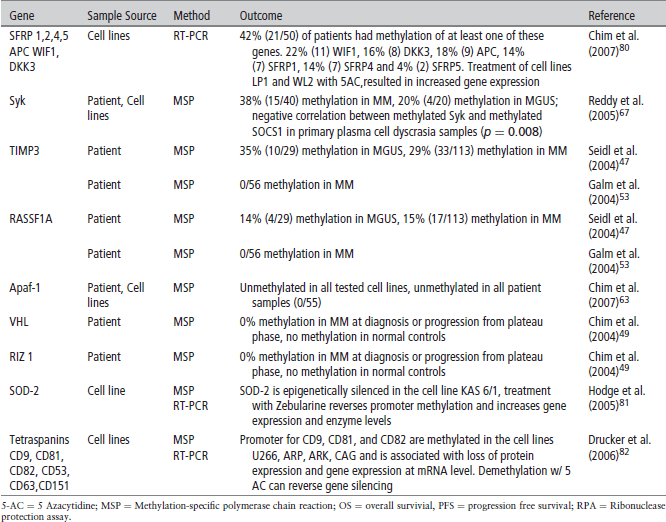
A recent study demonstrated the utility of examining sets of genes rather than single genes in myeloma pathogenesis. The investigators focused on the WNT pathway that is essential in embryogenesis, hematopoesis, and development. In the absence of WNT ligands, the protein, Beta-catenin, is phosphorylated leading to its own degradation. In the presence of Wnt ligand, there is hypophosphorylation and stabilization of β-catenin, allowing it to translocate to the nucleus and activate transcriptional events related to cell proliferation and survival. The WNT pathway can be inhibited by Wnt inhibitory factor 1 (WIF1), secreted frizzled-related proteins (SFRPs) and the Dickkopf (Dkk) family of secreted proteins. When looking at the methylation of each gene specifically, only a small percentage of patients show hypermethylation of the genes encoding these ligands, but when looking at various genes in the pathway, 42% of patients showed methylation and silencing of at least one of the seven genes examined in myeloma.80 This demonstrates the importance of analyzing multiple genes at once.
Investigators have also used transcriptional profiling after exposure to DNMT inhibitors as an indirect assay for uncovering epigenetically silenced genes. In one study, total cell RNA transcripts were evaluated before and after zebularine treatment and multiple genes were found to be upregulated in myeloma after treatment. This study found genes involved in apoptosis (BAD, BAK, BIK, and BAX) to be suppressed in KAS-6/1 cells and implicated them as a target of epigenetic silencing in myeloma.92
A better approach is to directly examine genome-wide methylation. This can be carried out by using several different methods:
(1) Capture bisulphite sequencing93 combines array-based hybrid selection and massively parallel bisulfite sequencing to profile DNA methylation. It is major limitation is the incomplete conversion of unmethylated cytosine to uracil resulting in false positive results for methylation.
(2) Methylated DNA Immunoprecipitation (MeDIP)94 uses antibodies against methylated DNA to enrich for the methylated DNA sequences, which are further analyzed using a microarray. This method has a relative poor specificity and is very dependent on CpG density.
(3) Comprehensive high-throughput arrays for relative methylation (CHARM)95 was recently published and takes advantage of the digestion of methylated DNA using McrBC followed by hybridization to a novel (CHARM) array. The authors furthermore developed new computational algorithms which average information from neighboring genomic locations.
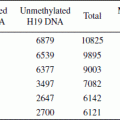
(4) The HpaII-tiny fragment enrichment by ligation mediated polymerase chain reaction (PCR) method (HELP assay),96 which was developed by our group. The HELP assay provides high-resolution analysis of cytosine methylation of DNA across the genome. This method is based on comparative MspI and HpaII isoschizomer digestion, size-specific amplification of DNA fragments and co-hybridization to custom high-density oligonucleotide arrays designed specifically for this method (Figure 1). HpaII is unable to digest methylated CpG at CCGG locations across the genome, whereas MspI is methylation-insensitive and can cut the same exact sites irrespective of methylation status. Thus, analyses of ratios of resulting different-sized fragments provide methylation data of uniform quality throughout the genome. We have demonstrated previously that HELP yields quantitative, reproducible (R < 0.98) and robust methylation signatures in cell lines, murine cells, as well as human normal and leukemia patient samples, as shown by bisulfite pyrosequencing validation.96 To make this assay even more comprehensive, a next generation array testing 1.13 million HpaII fragments has recently been developed that interrogates CCGG methylation status not just around promoter regions but also in coding sequences, introns, and intergenic regions. Many of those regions have recently been found to be very important for transcriptional and epigenetic regulation as they harbor distal regulatory elements, enhancers, or insulators that are critical for appropriate gene expression. In a further modification of this powerful method, this assay has been optimized to work with only 10 ng of total genomic DNA, thus making it useful for studies with primary tumor samples.
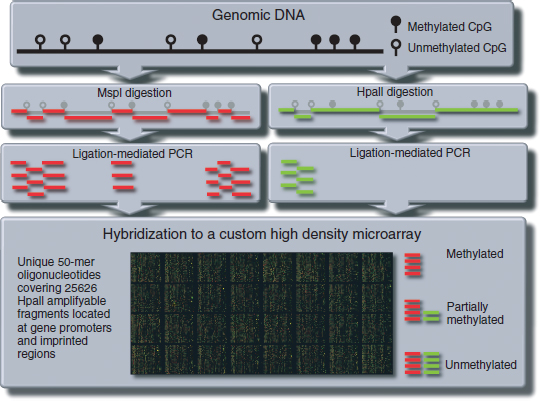
FIGURE 1 | The HELP (HpaII-tiny fragment enriched ligation PCR) assay. Genomic DNA is digested with HpaII and MspI restriction enzymes. Both enzymes recognize the same codon but HpaII will only cleave the DNA strand if the cytosine in the codon is methylated, whereas MspI will do so regardless of the methylation status. Restriction fragments are then amplified using a ligation mediated PCR with the PCR conditions set to amplify fragments between 200 and 2000 bp length. The amplification products are then labeled with a fluorochrome marker and are hybridized to a custom high-density oligonucleotide microarray.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


