Among individuals diagnosed with bone or soft tissue sarcoma, approximately 10% will have metastatic disease at the time of presentation,1–6 and an estimated 40% will develop metastatic disease during long-term follow-up.7,8 One of the historical challenges associated with managing patients with metastatic bone and soft tissue sarcoma has been the relative rarity of this disease: bone and soft tissue sarcoma represent only 1.1% of adult and 11.8% of pediatric malignancies in the developed world.9 Moreover, bone and soft tissue sarcomas do not behave as a single clinical entity, but actually comprise a heterogeneous group of over 50 distinct malignancies,10 each with important distinguishing features.
Management of patients with metastatic sarcoma is consequently best undertaken as a multidisciplinary effort, with input from pathology, radiology, radiation oncology, and medical and surgical subspecialties. Studies have routinely showed that these patients are best managed within a sarcoma specialty center.11,12 Although most patients with metastatic sarcoma are not treated with curative intent, selected patients with favorable histologies and clinical circumstances may experience long-term durable remissions with aggressive multidisciplinary management.
It is important for the practicing oncologist to be familiar with the natural history and the spectrum of management strategies for patients with metastatic disease.
Among patients with localized sarcoma, factors associated with risk of metastasis and disease-specific mortality in large registry studies are patient age, anatomic site of primary tumor, tumor size, tumor depth (superficial or deep to fascia), and histologic grade.13,14 The risk of metastasis appears most strongly correlated with tumor grade, with low-grade soft tissue sarcoma associated with a 10% to 20% risk of metastatic disease, compared to 40% to 50% for high-grade soft tissue sarcoma.15–17 The 7th edition of the American Joint Committee on Cancer (AJCC) Staging Manual for soft tissue sarcoma incorporates several of these factors, including tumor size (≤5 vs. >5 cm), depth (superficial or deep to fascia), regional nodal involvement, and FNCLCC histologic grade, as key components in the staging of patients with soft tissue sarcoma. Staging for bone sarcomas is similar, incorporating tumor size (≤8 vs. >8 cm), regional nodal involvement, and histologic grade.18 The general utility of the AJCC TNM staging system for predicting risk of disease-specific survival has been largely validated.13
However, tumor size, depth, and grade alone cannot fully describe a patient’s risk of metastasis. In particular, tumor histology has long been known to be important in determining risk and pattern of metastases: for example, large (>10 cm), well-differentiated liposarcomas of the extremities rarely metastasize, whereas large (>10 cm), pure myxoid liposarcomas of the trunk or extremities can metastasize in up to 30% of cases.19–22 In an attempt to address the importance of histologic subtype, Kattan et al included fibrosarcoma, leiomyosarcoma, liposarcoma, malignant fibrous histiocytoma (undifferentiated pleomorphic sarcoma), malignant peripheral nerve sheath tumors, and synovial sarcoma in their post-operative nomogram for estimation of disease-specific survival following primary resection of soft tissue sarcoma or recurrent soft tissue sarcoma.23,24 Several other groups have further extended this work, and histology-specific nomograms now exist for uterine leiomyosarcoma,25 synovial sarcomas,26 rhabdomyosarcomas,27 and several other histologic subtypes.28
In addition to histologic subtype, a novel algorithm based on the expression of genes related to mitosis and chromosomal maintenance (CINSARC) has been shown to correlate strongly with risk of metastasis, and improves on models based solely on FNCLCC grade.29 Incorporation of this, and other genomic biomarkers into existing sarcoma nomograms, will likely continue to aid in the refinement of our understanding of risk of metastasis.
Unlike carcinomas, where lymph node metastases are the most common initial site of metastasis, lymphatic spread is relatively uncommon in sarcomas, occurring in only 3% to 4% of all patients.30–32 Instead, hematogenous dissemination predominates, and circulating tumor cells can frequently be detected, even in early stages of disease.33–35 In a large EORTC study of 2185 patients, it was found that lung, liver, and bone are the most common sites of metastatic diseases, present in 54%, 19%, and 11% of patients with advanced sarcomas, respectively.36 As a general rule of thumb, anatomic site of the primary disease is a useful guide to likely patterns of metastasis, with patients with bone sarcoma and soft tissue sarcoma of the trunk and extremity being predisposed to develop lung metastases, and patients with visceral and abdominal primaries much more likely to develop liver metastases as the initial site of metastatic disease.17,37 Although the cumulative incidence of brain metastases is relatively low, occurring in 2.9% of patients with bone sarcomas and 3.5% of patients with soft tissue sarcomas,38 it can be much higher in individual histologic subtypes, approaching 40% in alveolar soft-part sarcoma, extraskeletal Ewing sarcoma, and hemangiopericytoma.39,40 Despite the relatively low incidence of nodal metastases in sarcomas in general, higher rates of nodal metastases have been reported in angiosarcomas, clear-cell sarcoma, epithelioid sarcoma, synovial sarcoma, and rhabdomyosarcoma.32,41,42 For these subtypes, sentinel node biopsy and/or elective lymph node dissection may be considered based on institutional experience and expertise.43,44
All patients with a new diagnosis of sarcoma should be evaluated by a multidisciplinary team familiar with the management of patients with sarcoma. Patients should have a complete history and physical examination and routine blood counts and chemistries to establish baseline performance status and physiologic reserve. In addition to imaging of the primary tumor, radiographic staging studies for patients with newly diagnosed sarcoma should be based on likely patterns of metastatic spread. Spiral computed tomography (CT) imaging of the chest appears to have the highest sensitivity for the detection of pulmonary metastases, whereas nuclear imaging with PET/CT has been found to have high sensitivity and specificity for the detection of extrapulmonary disease in patients with bone and soft tissue sarcoma.45,46 MRI imaging of the complete spine may benefit patients with myxoid and round cell liposarcomas, and MRI imaging of the brain should be considered in histologic subtypes with a predilection for brain metastases.
It is important to note that radiographic imaging may not reliably distinguish between benign disease and malignancy for lesions smaller than 1 cm in maximum diameter. For these lesions, serial imaging or tissue sampling may be required to establish or exclude a diagnosis of metastatic sarcoma. For patients with radiographic suggestion of possible distant disease, biopsy-confirmation of metastasis is preferred to exclude benign disease or a second primary malignancy, prior to formulation of a final treatment plan.
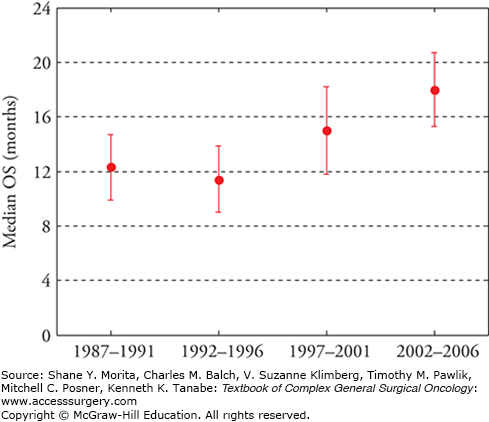
The overall prognosis for patients with metastatic sarcoma remains poor. However, considerable gains have been made in recent years, with several studies suggesting that median survival has improved by roughly 50% in the last 20 years, and is now estimated at 18 months, with 2-year overall survival nearing 40% (Fig. 30-1).7,36,47,48 Major changes driving improvements in cancer-specific survival have been the increased adoption of histology-specific chemotherapeutic regimens including the adoption of targeted therapies for selected patients; increased emphasis on a multimodality approach with integration of medical, surgical, and interventional approaches; and improvements in supportive care. Major considerations in determining the optimal treatment plan for metastatic sarcoma include the following:
FIGURE 30-1
Trends in overall survival among patients diagnosed with metastatic, non-GIST soft tissue sarcoma over the period from 1987 to 2006 as reported by the French Sarcoma Group Database. Median overall survival (OS) has improved roughly 50% from the initial study period (1987–1991, median OS = 12.3 months) to the most recent study period (2002–2006, median OS = 18 months). (Data from Italiano A, Mathoulin-Pelissier S, Cesne AL, et al. Trends in survival for patients with metastatic soft-tissue sarcoma, Cancer. March 1, 2011;117(5):1049–1054.)
Chemosensitivity of the histologic subtype
Time from diagnosis of localized disease to diagnosis of metastatic disease
Anatomic site(s) of metastasis and feasibility of (complete) surgical resection
Patient performance status
Major treatment modalities are outlined below.
For most patients with metastatic sarcoma, systemic chemotherapy is an appropriate first-line approach, particularly for individuals with multifocal disease and for patients with relatively chemosensitive histologies. Since the introduction of methotrexate in the late 1940s, there has been measured progress in the development and use of chemotherapy for the treatment of metastatic sarcoma. In addition to the development of novel drugs, there has also been an increased recognition of the fact that sarcomas are a heterogeneous group of distinct tumors, and treatments should be tailored to individual histologies and molecular aberrations if possible.
Common chemotherapeutic agents used in the treatment of bone and soft tissue sarcoma area are summarized in Table 30-1, and selected histology-specific regimens are outlined in Table 30-2.
Common Cytotoxic Chemotherapeutic Agents used in Sarcoma
| Drug | Class | Selected Toxicities |
|---|---|---|
| Cisplatin | Heavy metal, alkylating | Myelosuppression, nephrotoxicity, ototoxicity, neuropathy |
| Cyclophosphamide | Alkylating agent | Myelosuppression, hemorrhagic cystitis |
| Dacarbazine | Alkylating-like agent | Myelosuppression, gastrointestinal toxicity |
| Docetaxel | Taxane | Myelosuppression, peripheral neuropathy |
| Doxorubicin | Anthracycline | Myelosuppression, cardiotoxicity, mucositis |
| Etoposide | Topoisomerase II inhibitor | Myelosuppression |
| Gemcitabine | Antimetabolite | Myelosuppression |
| Ifosfamide | Alkylating agent | Myelosuppression, cystitis |
| Irinotecan | Topoisomerase I inhibitor | Myelosuppression, diarrhea |
| Methotrexate | Antimetabolite | Myelosuppression, mucositis, hepatotoxicity |
| Paclitaxel | Taxane | Myelosuppression, peripheral neuropathy |
| Temozolomide | Alkylating-like agent | Myelosuppression |
| Trabectedin | Marine-derived, alkylating | Myelosuppression, hepatotoxicity |
| Vincristine | Vinca alkyloid | Peripheral neuropathy |
| Vinorelbine | Vinca alkyloid | Myelosuppression, peripheral neuropathy |
Selected Histology-Specific Chemotherapeutic Regimens in Bone and Soft Tissue Sarcoma
| Histology | Regimen | References |
|---|---|---|
| Alveolar soft-part sarcoma (ASPS) | Sunitinib 37.5 mg daily | 117 |
| Angiosarcoma | Paclitaxel 80 mg/m2 d1,8,15 q28d | 75 |
| Bevacizumab 15 mg/kg q21d | 163 | |
| Clear-cell sarcoma | Sunitinib 37.5 mg daily | 115 |
| Tivantinib 360 mg bid | 164 | |
| Chondrosarcoma, high-grade | Cyclophosphamide 100 mg bid d1-7,15-21 + sirolimus 1 mg tid d1-21 q28d | 165 |
| Chordoma | Imatinib 800 mg daily | 109 |
| Lapatinib 1500 mg daily | 166 | |
| Desmoid | Sulindac 300 mg + tamoxifen 120 mg daily | 167 |
| Methotrexate 30 mg/m2 + vinblastine 5 mg/m2 weekly for 26 weeks, then every other week | 168 | |
| Sorafenib 400 mg daily | 169 | |
| Dermatofibrosarcoma protuberans (DFSP) | Imatinib 400 mg bid or 600 mg daily | 106,107 |
| Ewing sarcoma | VAC/IE (vincristine, doxorubicin + cyclophosphamide alternating with ifosfamide + etoposide) | 56 |
| Gastrointestinal stromal tumor (GIST) | Imatinib 400 mg daily or bid | 103,104 |
| Sunitinib 50 mg daily d1-28 every 6 weeks | 112 | |
| Regorafenib 160 mg daily | 119 | |
| Giant-cell tumor of bone | Denosumab 120 mg q28d | 123 |
| Inflammatory myofibroblastic tumor (IMT) | Crizotinib 250 mg bid | 170 |
| Osteosarcoma | MAP (high-dose methotrexate, doxorubicin, and cisplatin) | 171 |
| PEComa | Sirolimus 0.25 mg/m2/d, adjusted for target blood sirolimus level of 1-5 ng/mL | 172 |
| Everolimus 10 mg daily | 173 | |
| Rhabdomyosarcoma, nonpleomorphic | VAC (vincristine, doxorubicin, and cyclophosphamide) | 174 |
| Solitary fibrous tumor | Temozolomide 150 mg/m2 d1-7,15-21 + bevacizumab 5 mg/kg d8,22 q28d | 175 |
| Sunitinib 37.5 mg daily | 116 |
Conventional cytotoxic chemotherapeutic agents, which comprise the majority of drugs currently used in the treatment of sarcoma, are most toxic to rapidly dividing cells. Common mechanisms of action for these agents are the inhibition of DNA replication, direct damage to DNA, and disruption of the mitotic spindle cell apparatus. Several representative drugs pertinent to sarcoma treatment are summarized below.
Doxorubicin belongs to the class of anthracycline chemotherapeutic drugs and forms the backbone of many current regimens for bone and soft tissue sarcoma. Its primary antineoplastic mechanism of action is via DNA damage through topoisomerase II inhibition and direct cellular oxidative stress.49 Efficacy of doxorubicin against metastatic sarcomas was first demonstrated in the 1970s.50 Response rates for single-agent doxorubicin in all sarcoma histologies appear to be dose-dependent, with objective responses of 18%, 20%, and 37% in patients treated with escalating doses of 45, 60, and 75 mg/m2 administered intravenously every 21 days, respectively.51 A unique toxicity of doxorubicin is the risk for cardiomyopathy, particularly with cumulative lifetime doses exceeding 450 mg/m2. Swain and colleagues52 reported overt clinical heart failure in 0.2%, 1.6%, 3.3%, and 8.7% of patients receiving 150, 300, 450, and 600 mg/m2 of doxorubicin, respectively. Beyond limiting lifetime doxorubicin doses, treatment with dexrazoxane, beta-blockers, statins, and angiotensin inhibitors, which is associated with reduced cardiac risk, should be considered in patients at risk for anthracycline-mediated cardiotoxicity, particularly those patients with high cumulative lifetime doses.53 A liposomal formulation of doxorubicin is also available, which has a unique tissue distribution when compared to standard doxorubicin, though its therapeutic efficacy in all sarcoma subtypes has not been clearly defined.
Cyclophosphamide, one of the earliest alkylating agents, is an oxazaphosphorine-type agent that exerts its effects by cross-linking tumor DNA. Response rates of roughly 9% have been observed when used as a single agent in adult soft tissue sarcomas.54 It is now rarely used as a single agent for the treatment of bone and soft tissue sarcoma, but it is considered standard of care when used in combination regimens for the treatment of rhabdomyosarcoma and Ewing family sarcomas. It is also frequently used in relapsed/refractory osteosarcomas.55–57 Ifosfamide is an analog of cyclophosphamide that was introduced in the 1960s as a potentially more effective alkylating agent than cyclophosphamide. Initial studies demonstrated modest activity as a single agent both as first-line treatment and in refractory bone and soft tissue sarcomas, with a response rate of 18% when administered at a dose of 5 g/m2.54 In this early study, ifosfamide was superior to cyclophosphamide in terms of response rate and tolerability. Bolus dosing of ifosfamide appears to be associated with improved responses when compared with administration as a continuous infusion; a study at Dana Farber Cancer Institute documented response rates of 9% with continuous infusions of ifosfamide given at 2 g/m2/day over 4 days compared with a response rate of 26% in patients treated with 2 g/m2/day administered as bolus infusions over 4 hours on four consecutive days.58 Higher doses also appear to be associated with increased response rates, and doses up to 14 g/m2 per cycle have been shown to be feasible, albeit at the cost of increased toxicities.59 During initial drug development, hemorrhagic cystitis, caused by accumulation of the ifosfamide metabolite acrolein in the bladder, was the major dose-limiting toxicity; however, this has been largely mitigated by the coadministration of mesna as a chemopreventative together with aggressive hydration.60 Cytopenias and central nervous system (CNS) toxicity (delirium) remain important dose-limiting toxicities. Synovial sarcomas appear to be uniquely sensitive to ifosfamide, and treatment with an ifosfamide-containing regimen has been associated with significant improvements in progression-free survival.61
Gemcitabine is a nucleoside analog that is incorporated into replicating DNA, inhibiting cell division, and potentiating tumor cell death.62 When administered as first-line therapy or as second-line therapy to unselected patients with bone or soft tissue sarcoma refractory to doxorubicin- or ifosfamide-based chemotherapy, responses have been relatively low (3% to 7%).63–66 However, angiosarcomas may be uniquely sensitive, with one retrospective study reporting responses in two-thirds of patients with angiosarcoma treated with weekly gemcitabine.67 Despite the relatively low incidence of objective response in most sarcoma subtypes, gemcitabine is relatively well tolerated, and has synergy with several other chemotherapeutic agents. In particular, gemcitabine-based regimens incorporating dacarbazine and docetaxel are widely used in the treatment of metastatic sarcomas.
The mechanism of action of dacarbazine is not fully characterized, but it is believed to act both as an alkylating agent and as a purine analog to inhibit DNA synthesis. It has similar efficacy as doxorubicin and ifosfamide in unselected patients with soft tissue sarcoma when administered as a single agent at doses of 1200 mg/m2 every 3 weeks, with reported objective responses of 18%.68 The primary toxicity is hematologic, and anemia, thrombocytopenia, and granulocytopenia are dose-limiting. Dacarbazine is frequently used in combination with other agents, including doxorubicin, ifosfamide, and gemcitabine, in the treatment of soft tissue sarcoma.
Cisplatin belongs to the platinum group of chemotherapeutic agents. Direct DNA damage by cross-linking appears to be a critical determinant of its antineoplastic effect, though several recent studies also suggest that cisplatin can alter cellular metabolism and oxidative phosphorylation leading to cancer cell apoptosis.69 Cisplatin has a major role in the treatment of osteosarcoma as part of a multidrug combination therapy, commonly administered with doxorubicin in sequence with high-dose methotrexate. Major dose-limiting toxicities for cisplatin include emetogenicity, neuropathy, and nephrotoxicity. Severe peripheral neuropathy may be seen in patients with high cumulative doses, with roughly 40% of patients developing neuropathy with lifetime doses exceeding 300 mg/m2.70 Patients should undergo an otologic screening evaluation prior to initiation of cisplatin and be closely monitored for symptoms of ototoxicity during therapy, given the risk of irreversible high-frequency hearing loss.
Paclitaxel was initially derived from the bark of the Pacific yew tree, Taxus brevifolia, and exerts its primary antineoplastic effect by stabilizing microtubules and interfering with the function of the mitotic spindle complex.71 In unselected patients with bone and soft tissue sarcoma, paclitaxel was considered relatively inactive, with reported response rates of only 7%.72 However, angiosarcomas were noted to be particularly responsive, and two subsequent retrospective studies of paclitaxel in angiosarcoma reported response rates of up to 53% to 62%.73,74 Objective responses were somewhat lower (18%) in a prospective study of weekly paclitaxel at a dose of 80 mg/m2, though treatment was generally considered well tolerated.75 Major toxicities include hypersensitivity reactions, peripheral neuropathy, and cytopenias with prolonged administration. Docetaxel is a close analog of paclitaxel and shares its mechanism of action. Initial studies showed that docetaxel has activity in advanced soft tissue sarcomas, with responses of 17% as a single-agent when administered at a dose of 100 mg/m2 every 3 weeks in patients with otherwise chemorefractory disease.76 Docetaxel is frequently used in combination with gemcitabine for the treatment of bone and soft tissue sarcoma.
Vincristine and vinblastine are the two most widely used drugs in the class of vinca alkaloids, derived from the periwinkle plant, Catharanthus roseus. Vinca alkaloid chemotherapeutic agents, similar to taxanes, exert their primary effect by interfering with normal microtubule cellular dynamics. Although taxanes bind and stabilize existing microtubules, vinca alkaloids bind to tubulin monomers and prevent polymerization and formation of new microtubules, leading to metaphase arrest. Vincristine forms the backbone of treatment in the rhabdomyosarcoma and Ewing-family tumors. Major toxicities include peripheral neuropathy, constipation, and bone marrow suppression.
Methotrexate is an antifolate chemotherapy agent first developed in the 1940s to exploit the dependence of several cancer types on folate for DNA and RNA replication and repair.77 Methotrexate directly inhibits the enzyme dihydrofolate reductase, leading to the intracellular accumulation of the toxic metabolite tetrahydrofolate polyglutamate. Conventional osteosarcomas appear to be uniquely sensitive to methotrexate, particularly at high doses up to 20 g/m2.78 When administered at high doses (>1 g/m2), methotrexate should be administered with the fully reduced folate coenzyme, leucovorin, to protect against severe systemic toxicity. Key dose-limiting toxicities include cytopenias, mucositis, and risk of hepatic fibrosis and acute pneumonitis. Methotrexate has been used as a single agent for nonpleomorphic rhabdomyosarcoma, particularly for patients with high risk of CNS disease,79 but it is more commonly incorporated into multiagent chemotherapy for conventional osteosarcoma.80
Trabectedin is a novel chemotherapeutic drug derived from the marine tunicate (“sea squirt”), Ecteinascidia turbinata, that binds to the DNA minor groove, leading to interference with several DNA binding proteins and inhibition of DNA excision repair.81 Early work with this agent suggested in vitro efficacy against sarcoma cell lines. Subsequent studies have suggested activity in soft tissue sarcoma, in particular myxoid liposarcoma and uterine leiomyosarcomas.82–86 Trabectedin was approved by the European Commission in 2007 for treatment of patients with advanced soft tissue sarcoma on the basis of these studies. Important dose-limiting toxicities appear to be cytopenias and elevations in liver transaminases.
Thanks in large part to large multi-institution studies in pediatric and young adult patients, combination chemotherapy with a doxorubicin-containing regimen is now considered standard of care for patients with selected pediatric-type sarcomas, including metastatic osteosarcoma, Ewing sarcoma, and rhabdomyosarcoma. The role of doxorubicin-based combination chemotherapy is a bit more nuanced for most other patients with metastatic soft tissue sarcomas. A number of trials have compared doxorubicin monotherapy with doxorubicin-based combination chemotherapy in adult patients with advanced soft tissue sarcoma.87–92 Addition of ifosfamide or dacarbazine to doxorubicin is associated with better radiographic response rates; however, multidrug regimens have also been associated with added toxicity, and no differences in overall survival. A meta-analysis of three randomized phase 3 clinical trials evaluating the impact of adding ifosfamide to doxorubicin-based regimens showed a roughly 50% improvement in objective responses, but no difference in overall survival at 1 year with the combination regimen.93 However, addition of ifosfamide to doxorubicin in the adjuvant setting following complete resection may be associated with improvements in overall recurrence risk, as reported in a recent meta-analysis.94 Consequently, most clinicians would reserve multidrug doxorubicin-based chemotherapy for (1) patients with relatively chemosensitive histologies, in which combination therapy is considered the standard of care, (2) patients with a good performance status who are symptomatic from their disease and who would likely derive symptomatic benefit from tumor shrinkage, and (3) those patients who are candidates for complete surgical resection of their disease.95 Combination therapy may also be considered on a case-by-case basis for patients with an excellent baseline performance status.
Though gemcitabine has limited activity as a single agent in sarcoma, it appears to have synergistic activity when used in combination with other agents. Several combination regimens are of interest. In a large study by the Sarcoma Alliance for Research through Collaboration (SARC), 122 patients with metastatic soft tissue sarcoma were randomized to single-agent gemcitabine or combination chemotherapy with gemcitabine and docetaxel. Among patients treated with the combination regimen, there were improvements in objective responses (16% vs. 8%), progression-free survival (6.2 vs. 3 months), and overall survival (17.9 vs. 11.5 months).96 This regimen appears to have activity in multiple sarcoma subtypes, but may be uniquely active in patients with uterine leiomyosarcoma. Hensley et al reported their experience with patients with metastatic leiomyosarcoma, primarily of uterine origin, who had failed at least one prior systemic chemotherapy regimen. Objective responses were seen in 53% of patients, including complete responses in 9% of patients.97 Two prospective studies conducted by the Gynecologic Oncology Group (GOG) on uterine leiomyosarcoma showed objective responses in 36% of patients treated with gemcitabine and docetaxel as first-line therapy, and in 27% of patients treated with the combination regimen as second-line therapy.98,99
Garcia-Del-Muro and colleagues100 evaluated gemcitabine administered at a dose of 1800 mg/m2 every 2 weeks in combination with dacarbazine 500 mg/m2 in comparison with standard single-agent dacarbazine at 1200 mg/m2 every 21 days in pretreated patients with soft tissue sarcoma. Combination therapy was associated with statistically significant improvements in progression-free survival at 3 months (56% vs. 37%) and median overall survival (16.8 vs. 8.2 months). Importantly, combination therapy was better tolerated than single-agent dacarbazine, with fewer treatment delays observed with combination therapy.
Identification of molecular drivers of sarcomagenesis and potential targets for novel therapeutic agents has been a challenging endeavor, in part due to the heterogeneity of sarcomas as well as the rarity of these tumors in comparison to other cancers. However, there have been several notable successes. Molecular studies of gastrointestinal stromal tumors (GISTs) in the 1990s led to the identification of activating mutations in the proto-oncogenes KIT and PDGFRA in the majority of patients with GIST, motivating the evaluation of imatinib, an oral inhibitor of KIT and PDGFRA, as a potential therapeutic agent.101 This represented the first time that a druggable driver mutation had been identified in bone and soft tissue sarcoma. Subsequent molecular studies have identified other potentially druggable mutations in a number of sarcomas. A selected list of targets is outlined in Table 30-3.
Selected Molecular Targets in Sarcoma
| Target(s) | Relevant Histologic Subtype(s) | Selected Drug(s) | Reference(s) |
|---|---|---|---|
| ALK | Inflammatory myofibroblastic tumor (IMT) | Crizotinib | 170 |
| CSF1, CSF1R | Pigmented villonodular synovitis (PVNS), giant-cell tenosynovitis | Imatinib | 108 |
| EGFR | EGFR-positive chordomas | Erlotinib, lapatinib | 166,176 |
| KIT, PDGFR | GIST | Imatinib, sunitinib, regorafenib | 103,104,112,119 |
| HER2/neu | HER2-positive osteosarcoma | Trastuzumab | |
| IGF1R | Ewing family of tumors | Cixutumumab, figutumumab, ganitumab | 177–179 |
| MDM2, CDK4 | Well-differentiated and dedifferentiated liposarcoma | RG7112, PD0332991 | 180,181 |
| MET | Clear-cell sarcoma | Tivantinib | 164 |
| MTOR | PEComa, angiomyolipoma, lymphangioleiomyomatosis | Sirolimus, everolimus | 172,173 |
| PARP | Ewing sarcoma | Olaparib, veliparib | 182 |
| PDGFR | Dermatofibrosarcoma protuberans | Imatinib | 106,107 |
| RANK, RANKL | Giant-cell tumor of bone | Denosumab | 123 |
| TEM-1 | Soft tissue sarcoma | MORAb-004 | |
| VEGF, VEGFR | Angiosarcoma, epithelioid hemangioendothelioma, nonadipocytic soft tissue sarcomas | Bevacizumab, pazopanib, sunitinib | 115–117,122,163,175 |
| γ-secretase, Notch pathway | Desmoid tumors | RO4929097, PF-03084014 | 183 |
Imatinib is an oral tyrosine-kinase inhibitor with activity against multiple targets, including PDGFR, KIT, and CSF1R.102 Two large prospective studies of imatinib in patients with GIST showed that imatinib could induce radiographic responses in roughly half (45% to 52%) of all patients, in what was earlier considered a chemoresistant histology.103,104 Initial data suggested that treatment with a higher dose of 800 mg daily was associated with improved progression-free survival (PFS) when compared with 400 mg daily. However, a meta-analysis of available clinical trial data suggested that this improvement in PFS did not translate into an improvement in overall survival, given that dose escalation at progression could salvage a subset of these patients. Moreover, only patients with mutations in exon 9 of KIT appeared to derive a PFS benefit from initial higher doses of imatinib.105 Imatinib therapy, typically at a dose of 400 mg daily, is now considered first-line therapy for metastatic or unresectable GIST. Major toxicities of imatinib include fatigue, gastrointestinal symptoms, and cytopenias, particularly at higher doses.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


