Abbreviations
- ACTH Adrenocorticotropin
- ADHR Autosomal dominant hypophosphatemic rickets
- AHO Albright hereditary osteodystrophy
- AIRE Autoimmune regulator
- BMD Bone mineral density
- CaSR Extracellular calcium-sensing receptor
- CGRP Calcitonin gene–related peptide
- DBP Vitamin D–binding protein
- DXA Dual-energy x-ray absorptiometry
- FBHH Familial benign hypocalciuric hypercalcemia
- FGF23 Fibroblast-derived growth factor 23
- GALNT3 UDP-N-acetyl-α-D-galactosamine transferase
- HPT-JT Hyperparathyroidism-jaw tumor
- HT Hormone therapy
- ICMA Immunochemiluminescent assay
- IFN Interferon
- IGF Insulin-like growth factor
- IL Interleukin
- IP3 Inositol 1,4,5-triphosphate
- IRMA Immunoradiometric assay
- LDL Low-density lipoprotein
- MCT Medullary carcinoma of thyroid
- MEN Multiple endocrine neoplasia
- MEPE Matrix extracellular phosphoglycoprotein
- NALP5 NACHT leucine-rich-repeat protein 5
- OPG Osteoprotegerin
- PHP Pseudohypoparathyroidism
- PIP2 Phosphatidylinositol 4,5-bisphosphate
- PPHP Pseudo pseudohypoparathyroidism
- PTH Parathyroid hormone
- PTHrP Parathyroid hormone–related protein
- RANK Receptor activator of nuclear factor kappa B
- RANKL Receptor activator of nuclear factor kappa-B ligand
- RAR Retinoic acid receptor
- RBP Retinol-binding protein
- RXR Retinoid X receptor
- SERMs Selective estrogen response modulators
- sFRP Secreted frizzled related protein
- TNF Tumor necrosis factor
- TPN Total parenteral nutrition
- TRP Tubular reabsorption of phosphate
- VDR Vitamin D receptor
- VDRE Vitamin D response element
- VIP Vasoactive intestinal polypeptide
- WHI Women’s Health Initiative
- WHO World Health Organization
- XLH X-linked hypophosphatemia
Cellular and Extracellular Calcium Metabolism
The calcium ion plays a critical role in intracellular and extracellular events in human physiology. Extracellular calcium levels in humans are tightly regulated within a narrow physiologic range to provide for proper functioning of many tissues: excitation-contraction coupling in the heart and other muscles, synaptic transmission and other functions of the nervous system, platelet aggregation, coagulation, and secretion of hormones and other regulators by exocytosis. The level of intracellular calcium is also tightly controlled, at levels about 10,000-fold lower than extracellular calcium, in order for calcium to serve as an intracellular second messenger in the regulation of cell division, muscle contractility, cell motility, membrane trafficking, and secretion.
It is the concentration of ionized calcium ([Ca0<+]) that is regulated in the extracellular fluid. The ionized calcium concentration averages 1.25 ± 0.07 mmol/L (Table 8–1). However, only about 50% of the total calcium in serum and other extracellular fluids is present in the ionized form. The remainder is bound to albumin (about 40%) or complexed with anions such as phosphate and citrate (about 10%). The protein-bound and complexed fractions of serum calcium are metabolically inert and are not regulated by hormones; only the ionized [Ca2+] serves a regulatory role, and only this fraction is itself regulated by the calciotropic hormones parathyroid hormone (PTH) and 1,25 dihydroxyvitamin D [1,25(OH)2D]. Large increases in the serum concentrations of phosphate or citrate can, however, by mass action, markedly increase the complexed fraction of calcium. For example, massive transfusions of blood, in which citrate is used as an anticoagulant, can reduce the ionized [Ca2+] enough to produce tetany. In addition, because calcium and phosphate circulate at concentrations close to saturation, a substantial rise in the serum concentration of either calcium or phosphate can lead to the precipitation of calcium phosphate salts in tissues. This is a source of major clinical problems in patients with severe hypercalcemia (eg, malignant tumors) and in those with severe hyperphosphatemia (eg, in renal failure or rhabdomyolysis).
| Total serum calcium | 8.5-10.5 mg/dL (2.1-2.6 mmol/L) |
|---|---|
| Ionized calcium | 4.4-5.2 mg/dL (1.1-1.3 mmol/L) |
| Protein-bound calcium | 4.0-4.6 mg/dL (0.9-1.1 mmol/L) |
| Complexed calcium | 0.7 mg/dL (0.18 mmol/L) |
| Intracellular free calcium | 0.00018 mmol/L (180 nmol/L) |
What is remarkable about calcium metabolism is that the serum-ionized [Ca2+], which represents a tiny fraction of the total body calcium, can be so tightly regulated in the face of the rapid fluxes of calcium through body compartments that take place during the course of calcium metabolism (Figure 8–1). The total calcium in extracellular fluid amounts to about 1% of total body calcium, with most of the remainder sequestered in bone. Yet from the extracellular fluid compartment, which contains about 900 mg of calcium, 10,000 mg/d is filtered at the glomerulus and 500 mg/d is added to a labile pool in bone; and to the extracellular fluid compartment are added about 200 mg absorbed from the diet, 9800 mg reabsorbed by the renal tubule, and 500 mg from bone.
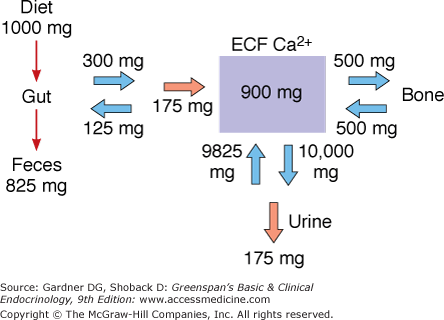
The challenge of calcium homeostasis, then, is to maintain a constant level of ionized [Ca2+] in the extracellular fluid, simultaneously providing adequate amounts of calcium to cells, to bone, and for renal excretion—and all the while compensating, on an hourly basis, for changes in daily intake of calcium, bone metabolism, and renal function. It is scarcely surprising that this homeostatic task requires two hormones, PTH and 1,25(OH)2D, or that the secretion of each hormone is exquisitely sensitive to small changes in the serum calcium, or that each hormone is able to regulate calcium exchange across all three interfaces of the extracellular fluid: the gut, the bone, and the renal tubule. We will reexamine the integrated roles of PTH and 1,25(OH)2D in calcium homeostasis after their actions and secretory control have been described.
The challenge of the cellular calcium economy is to maintain a cytosolic [Ca2+], or [Ca2+]i, of about 100 nmol/L, about 10,000-fold less than what is present outside cells (~1.0 mmol/L), providing for rapid fluxes through the intracellular compartment as required for regulation while maintaining a large gradient across the cell membrane. The calcium gradient across the cell membrane is maintained by ATP-dependent calcium pumps, Na+-Ca2+ exchangers, and the storage of calcium within intracellular sites. Calcium can enter cells through several types of calcium channels, some of which are voltage operated or receptor operated, to provide for rapid influx in response to depolarization or receptor stimulation. The cell also maintains large stores of calcium in microsomal and mitochondrial pools and in some cells the Golgi apparatus. Calcium can be released from microsomal stores rapidly by cellular signals such as 1,4,5-inositol trisphosphate (IP3). Reuptake mechanisms are also present, so that cytosolic calcium transients can be rapidly terminated by returning calcium to storage pools or pumping it across the plasma membrane.
PTH is secreted from four glands located adjacent to the thyroid gland in the neck. The glands weigh an average of 40 mg each. The two superior glands are usually found near the posterior aspect of the thyroid capsule; the inferior glands are most often located near the inferior thyroid margin. However, the exact location of the glands is variable, and 12% to 15% of normal persons have a fifth parathyroid gland. The parathyroid glands arise from the third and fourth branchial pouches. The inferior glands are actually those derived from the third branchial pouches. Beginning cephalad to the other pair, they migrate further caudad, and one of them sometimes follows the thymus gland into the superior mediastinum. The small size of the parathyroids and the vagaries of their location and number make parathyroid surgery a challenging enterprise for all but the expert surgeon.
The parathyroid glands are composed of epithelial cells and stromal fat. The predominant epithelial cell is the chief cell. The chief cell is distinguished by its clear cytoplasm from the oxyphil cell, which is slightly larger and has eosinophilic granular cytoplasm. Both cell types contain PTH, and it is not known whether their secretory regulation differs in any fundamental way.
To regulate the extracellular calcium concentration [Ca2+], PTH is under tight control by the serum calcium concentration. Thus, the negative feedback relationship of PTH with serum [Ca2+] is steeply sigmoidal, with the steep portion of the curve corresponding exactly to the normal range of serum calcium—precisely the relationship to create a high “gain” controller and ensure maintenance of the normal serum-ionized [Ca2+] by PTH (Figure 8–2).
Figure 8–2
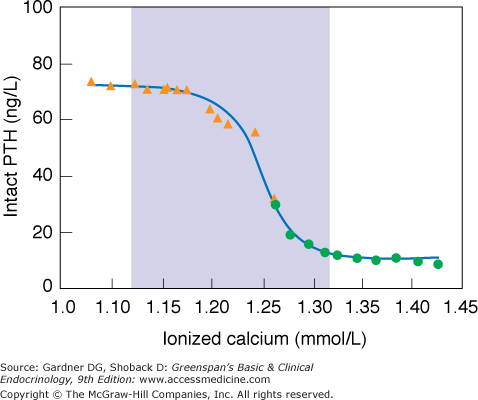
The relationship between the serum-ionized calcium level and the simultaneous serum concentration of intact PTH in normal humans. The serum calcium concentration was altered by the infusion of calcium (closed circles) or citrate (closed triangles). Parathyroid sensitivity to changes in serum calcium is maximal within the normal range (the shaded area). Low concentrations of PTH persist in the face of hypercalcemia.
(Modified from Conlin PR, et al. Hysteresis in the relationship between serum ionized calcium and intact parathyroid hormone during recovery from induced hyper- and hypocalcemia in normal humans. J Clin Endocrinol Metab. 1989;69:593. By permission of the Journal of Clinical Endocrinology and Metabolism.)
To sense the ionized [Ca2+] and thereby regulate the secretion of PTH, the parathyroid cell relies on relatively high levels of expression of the extracellular calcium-sensing receptor (CaSR). CaSR is a 120-kDa G protein–coupled receptor belonging to family C of this superfamily (see Chapter 1). The CaSR has sequence homologies to the metabotropic glutamate receptors of the central nervous system, the type B gamma-aminobutyric acid receptor, and a large family of pheromone receptors. The large extracellular domain of the CaSR mediates the sensing of calcium and other ions. Like other G protein–coupled receptors, the CaSR has seven membrane-spanning domains. The intracellular loops that connect these domains are directly involved in coupling the receptor to G proteins.
Shortly after the identification of the CaSR, it was shown that mutations in this receptor were responsible for familial benign hypocalciuric hypercalcemia (FBHH), a disorder of calcium sensing by the parathyroid glands and kidney. The CaSR is not unique to the parathyroid. CaSRs are widely distributed in the brain, skin, growth plate, intestine, stomach, C cells, and other tissues. This receptor regulates the responses to calcium in thyroid C cells, which secrete calcitonin in response to high extracellular [Ca2+], and in the distal nephron of the kidney, where the receptor regulates calcium excretion. The function of CaSRs in many other sites is still unclear.
The primary cellular signal by which an increased extracellular [Ca2+] inhibits the secretion of PTH appears to be an increase in [Ca2+]i. The CaSR is directly coupled by Gq to the enzyme phospholipase C, which hydrolyzes the phospholipid phosphatidylinositol 4,5-bisphosphate (PIP2) to liberate the intracellular messengers IP3 and diacylglycerol (see Chapter 1). IP3 binds to a receptor in endoplasmic reticulum that releases calcium from membrane stores. The release of stored calcium raises the [Ca2+]i rapidly and is followed by a sustained influx of extracellular calcium, through channels that produce a rise and sustained plateau in [Ca2+]i. Increased [Ca2+]i may be sufficient for inhibition of PTH release, but it is unclear whether calcium release from intracellular stores or sustained calcium influx from the cell exterior is more important. The other product of phospholipase C action is the lipid diacylglycerol, an activator of the calcium- and phospholipid-sensitive protein kinases in the protein kinase C family. The effects of protein kinase C isoenzymes on the release of PTH from the gland are complex. CaSRs also couple to the inhibition of cyclic adenosine-3′,5′-monophosphate (cAMP) generation, which also may play a role in setting the response of parathyroid cells to ambient calcium levels.
The initial effect of high extracellular calcium is to inhibit the secretion of preformed PTH from storage granules in the gland by blocking the fusion of storage granules with the cell membrane and release of their contents. In most cells, stimulation of exocytosis (stimulus-secretion coupling) is a calcium-requiring process, which is inhibited by depletion of extracellular calcium. The parathyroid cell is necessarily an exception to this rule, because this cell must increase secretion of PTH when the ionized [Ca2+] is low. In the parathyroids, intracellular magnesium may serve the role in stimulus-secretion coupling that calcium does in other cells. As discussed later in the section on hypoparathyroidism, depletion of magnesium stores can paralyze the secretion of PTH, leading to reversible hypoparathyroidism.
Besides calcium, there are several regulators of PTH secretion. Hypermagnesemia inhibits PTH, and during the treatment of premature labor with infusions of magnesium sulfate, reductions in PTH levels and occasionally hypocalcemia are observed. Conversely, moderate hypomagnesemia can stimulate PTH secretion, even though prolonged depletion of magnesium will paralyze it. On a molar basis, magnesium is less potent in controlling secretion than calcium. Catecholamines, acting through β-adrenergic receptors and cAMP, stimulate the secretion of PTH. This effect does not appear to be clinically significant. The hypercalcemia sometimes observed in patients with pheochromocytoma usually has another basis—secretion of parathyroid hormone–related protein (PTHrP) by the tumor.
Not only do changes in serum calcium regulate the secretion of PTH—they also regulate the synthesis of PTH at the level of stabilizing preproPTH mRNA levels. It is estimated that glandular stores of PTH are sufficient to maintain maximal rates of secretion for no more than 1.5 hours, so increased synthesis is required to meet sustained hypocalcemic challenges.
Transcription of the PTH gene is also regulated by vitamin D: high levels of 1,25(OH)2D inhibit PTH gene transcription. This is one of many ways that the calciotropic hormones cooperatively regulate calcium homeostasis, and it has therapeutic implications. Vitamin D analogs are used to treat secondary hyperparathyroidism in dialysis patients with renal osteodystrophy.
PTH is an 84-amino-acid peptide with a molecular weight of 9300. Its gene is located on chromosome 11. The gene encodes a precursor called preproPTH with a 29-amino-acid extension added at the amino terminus of the mature PTH peptide (Figure 8–3). This extension includes a 23-amino-acid signal sequence (the pre sequence) and a 6-residue prohormone sequence. The signal sequence in preproPTH functions precisely as it does in most other secreted protein molecules, to allow recognition of the peptide by a signal recognition particle, which binds to nascent peptide chains as they emerge from the ribosome and guides them to the endoplasmic reticulum, where they are inserted through the membrane into the lumen (Figure 8–4).
Figure 8–3
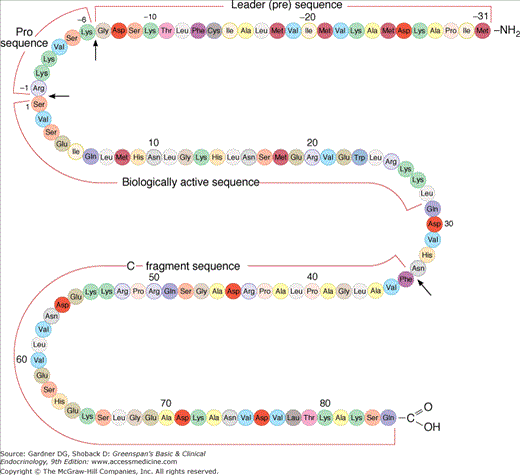
Primary structure of human preproparathyroid hormone. The arrows indicate sites of specific cleavages which occur in the sequence of biosynthesis and peripheral metabolism of the hormone. The biologically active sequence is enclosed in the center of the molecule.
(Reproduced, with permission, from Felig P, Baxter JD, Frohman LA, ed. Endocrinology and Metabolism. 3rd ed. McGraw-Hill; 1995.)
Figure 8–4
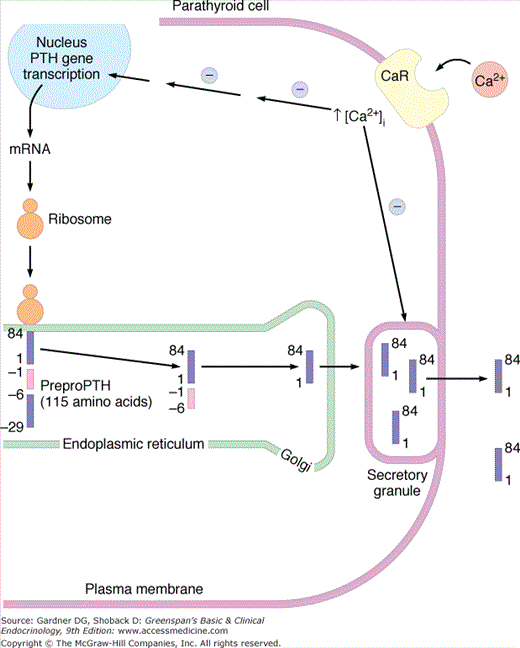
Biosynthetic events in the production of PTH within the parathyroid cell. PreproPTH gene is transcribed to its mRNA, which is translated on the ribosomes to preproPTH(amino acids −29 to +84). The presequence is removed within the endoplasmic reticulum, yielding proPTH(−6 to +84). Mature PTH(1-84) released from the Golgi is packaged in secretory granules and released into the circulation in the presence of hypocalcemia. The CaSR, or CaR, senses changes in extracellular calcium that affect both the release of PTH and the transcription of the preproPTH gene. (Reproduced, with permission, from McPhee SJ, et al, ed. Pathophysiology of Disease: An Introduction to Clinical Medicine. Originally published by Appleton & Lange. Copyright 1995 by The McGraw-Hill Companies, Inc.)
In the lumen of the endoplasmic reticulum, a signal peptidase cleaves the signal sequence from preproPTH to leave proPTH, which exits the endoplasmic reticulum and travels to the Golgi apparatus, where the pro sequence is cleaved from PTH by an enzyme called furin. Whereas preproPTH is evanescent, proPTH has a life span of about 15 minutes. The processing of proPTH is quite efficient, and proPTH, unlike other prohormones (eg, proinsulin), is not secreted. As it leaves the Golgi apparatus, PTH is repackaged into dense neuroendocrine-type secretory granules, where it is stored to await secretion.
PTH secreted by the gland has a circulating half-life of 2 to 4 minutes. Intact PTH(1-84) is predominantly cleared in the liver and kidney. There, PTH is cleaved at the 33 to 34 and 36 to 37 positions to produce an amino terminal fragment and a carboxyl terminal fragment. Amino terminal fragments of PTH do not circulate to the same extent as carboxyl terminal fragments. The latter are cleared from blood by renal filtration, and they accumulate in chronic renal failure. Although the classic activities of PTH are encoded in the amino terminal portion of the molecule, mid region and carboxyl terminal fragments of the hormone may not be metabolically inert. Recent evidence suggests that they may have their own receptors and biologic actions.
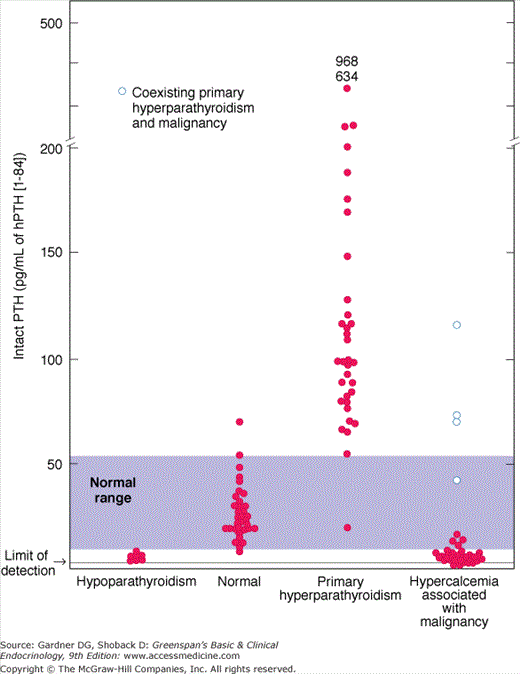
Current assays of intact PTH(1-84) employ two-site immunoradiometric assay (IRMA) or immunochemiluminescent assay (ICMA) techniques, in which the normal range for PTH is approximately 10 to 60 pg/mL (1-6 pmol/L). By utilizing antibodies to two determinants, one near the amino terminal end of the PTH molecule and the other near the carboxyl terminal end of PTH, these assays are designed to measure the intact, biologically active hormone species (Figure 8–5). In practice, such assays have sufficient sensitivity and specificity to detect not only increased levels of PTH in patients with hyperparathyroid disorders but also suppressed levels of PTH in patients with nonparathyroid hypercalcemia. The ability to detect suppression of PTH makes these assays powerful tools for the differential diagnosis of hypercalcemia. If hypercalcemia results from a form of hyperparathyroidism, then the serum PTH level will be high; if hypercalcemia has a nonparathyroid basis, then PTH will be suppressed.
Figure 8–5
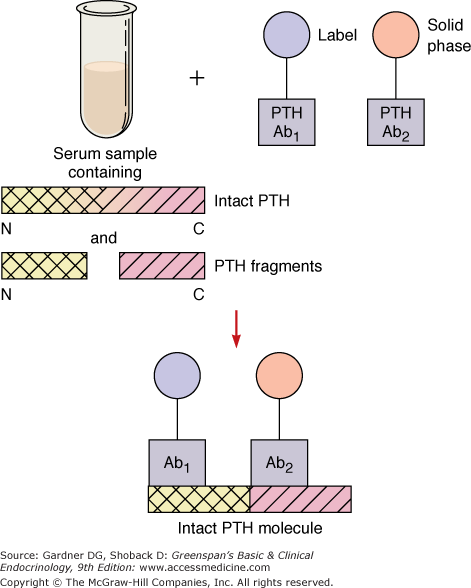
Schematic representation of the principle of the two-site assay for intact PTH. The label may be a luminescent probe or 125 I in the immunochemiluminescent or immunoradiometric assay, respectively. Two different region-specific antibodies are used (Ab1 and Ab2). Only the hormone species containing both immunodeterminants is counted in the assay. Most recent whole or intact PTH immunoassays employ an Ab1 directed against extreme N-terminal PTH epitopes as shown.
(Reproduced and modified, with permission, from McPhee SJ, et al, ed. Pathophysiology of Disease: An Introduction to Clinical Medicine. Originally published by Appleton & Lange. Copyright 1995 by The McGraw-Hill Companies, Inc.)
Recently, intact PTH assays have been further refined so that the amino-terminal antibody recognizes only the most amino terminal residues of PTH (essentially amino acids 1-6). This improvement was made because it became clear that antibodies used in first-generation, two-site intact PTH assays were detecting species of PTH that were not PTH (1-84). There are non-PTH(1-84) amino-terminally truncated PTH fragments generated in vivo by peripheral metabolism that can accumulate in patients with chronic renal failure. About 50% of circulating PTH in patients with renal failure consists of large fragments of non-(1-84)PTH. Assays specific for full-length PTH, in which the amino terminal antibodies truly detect residues at the extreme N-terminus of the molecule (residues 1-6), are designated as whole, bioactive, or intact PTH.
The central function of PTH is to regulate ionized [Ca2+] levels by concerted effects on three principal target organs: bone, intestinal mucosa, and kidney. The effect of PTH on intestinal calcium absorption is indirect, resulting from increased renal production of the intestinally active vitamin D metabolite 1,25(OH)2D. By its integrated effects on the kidney, gut, and bone, PTH acts to increase the inflow of calcium into the extracellular fluid and thus defend against hypocalcemia. Removal of the parathyroid glands results in profound hypocalcemia and ultimately in tetany and death.
In the kidney, PTH has direct effects on the tubular reabsorption of calcium, phosphate, and bicarbonate. Although the bulk of calcium is resorbed from tubule fluid together with sodium in the proximal convoluted tubule, the fine tuning of calcium excretion occurs in the distal nephron. There, PTH markedly increases the reabsorption of calcium, predominantly in the distal convoluted tubule. Although calcium is actively transported against an electrochemical gradient, the precise nature of the calcium transport process that is regulated by PTH is controversial. However, from a physiologic standpoint, the ability to limit renal losses of calcium is one important means by which PTH defends the serum calcium level.
PTH inhibits the reabsorption of phosphate in the renal proximal tubule. In this nephron segment, phosphate is transported across the apical membrane of the tubule cell by specific sodium-phosphate cotransporters, with phosphate influx driven by the energy of the sodium gradient. PTH inhibits sodium-phosphate reabsorption by reducing the rate of insertion of transporters from a sequestered cytoplasmic pool into the apical membrane. The phosphaturic effect of PTH is profound. It is best quantified by calculating the tubular reabsorption of phosphate (TRP) from the clearances of phosphate and creatinine (TRP = 1 − CP/Ccreat, normal range 80%-97%), or by calculating the renal phosphate threshold (TmP/GFR) from a standard nomogram. Because it is primarily the renal phosphate threshold that sets the level of serum phosphate, the phosphaturic effect of PTH is mirrored in the serum phosphorus level (eg, hypophosphatemia in hyperparathyroidism). Hyperparathyroid states may also be characterized by impaired bicarbonate reabsorption and a mild hyperchloremic metabolic acidosis, because of inhibition of Na+-H+ antiporter activity by PTH.
Although the hypocalciuric effect of PTH is readily understood as part of the concerted actions of the hormone to defend the serum [Ca2+], the utility of the phosphaturic effect of PTH is less obvious. One consideration is that the phosphaturic effect tends to prevent an increase in serum phosphate, which would otherwise result from the obligatory release of phosphate with calcium during bone resorption and would tend to dampen the homeostatic increase in serum calcium by complexing calcium in blood. An example is renal osteodystrophy. When phosphate clearance is impaired by renal failure, the hypocalcemic effect of phosphate released during bone remodeling is an important contributor to progressive secondary hyperparathyroidism as part of a positive feedback loop—the more that bone resorption is stimulated and phosphate released, the more hyperparathyroidism is induced.
There are two mammalian receptors for PTH. The first receptor to be identified recognizes PTH and PTHrP and is designated the PTH/PTHrP receptor, or the PTH-1 receptor (PTH-1R). The PTH-2R is activated by PTH only. These receptors also differ in their tissue distribution. The PTH-1R in kidney and bone is an 80,000-MW glycoprotein and member of the G protein receptor superfamily. It has the canonical architecture of such receptors, with a large first extracellular domain, seven consecutive membrane-spanning domains, and a cytoplasmic tail (see Chapter 1). PTH binds to sites in the large extracellular domain of the receptor. The hormone-bound form of the receptor then activates associated G proteins via several determinants in the intracellular loops. PTH receptors are closely related to a small subfamily of peptide hormone receptors, which includes those for secretin, vasoactive intestinal polypeptide (VIP), adrenocorticotropin (ACTH), and calcitonin.
PTH itself has a close structural resemblance to a sister protein, PTHrP, and also resembles the peptides secretin, VIP, calcitonin, and ACTH. As noted above, the receptors for these related ligands are themselves members of a special family. These peptide hormones are characterized by an amino terminal alpha helical domain, thought to be directly involved in receptor activation, and an adjacent alpha helical domain that seems to be the primary receptor-binding domain. In the case of PTH, residues 1 to 6 are required for activation of the receptor; truncated analogs without these residues (eg, PTH[7-34]) can bind the receptor but cannot efficiently activate it and thus serve as competitive antagonists of PTH action. The primary receptor-binding domain consists of PTH(18-34). Although the intact form of PTH is an 84-amino-acid peptide, PTH(35-84) does not seem to have any important role in binding to the bone-kidney receptor. However, a separate PTH(35-84) receptor may exist; the carboxyl terminal PTH receptor could mediate an entirely different set of actions of PTH.
The PTH-1 receptor binds PTH and PTHrP with equal affinity. Physiologic activation of the receptor by binding of either PTH or PTHrP induces the active, GTP-bound state of two receptor-associated G proteins. Gs couples the receptor to the effector adenylyl cyclase and thereby to the generation of cAMP as a cellular second messenger. Gq couples the receptor to a separate effector system, phospholipase C, and thereby to an increase in [Ca2+]i and to activation of protein kinase C (see Chapter 1). Although it is not clear which of the cellular messengers, cAMP, intracellular calcium, or diacylglycerol is responsible for each of the various cellular effects of PTH, there is evidence from an experiment of nature that cAMP is the intracellular second messenger for maintaining calcium homeostasis and renal phosphate excretion. The experiment is pseudohypoparathyroidism (PHP), in which null mutations in one allele of the stimulatory G protein subunit Gs alpha (GNAS1) cause hypocalcemia and unresponsiveness of renal phosphate excretion to PTH.
When secreted in abundance by malignant tumors, PTHrP produces severe hypercalcemia by activating the PTH-1 receptor. However, the physiologic roles of PTHrP are quite different from those of PTH. PTHrP is produced in many fetal and adult tissues. Based on gene knockout experiments and overexpression of PTHrP in individual tissues, we now know that PTHrP is required for normal development as a regulator of the proliferation and mineralization of chondrocytes and as a regulator of placental calcium transport. In postnatal life, PTHrP also appears to regulate epithelial-mesenchymal interactions that are critical for development of the mammary gland, skin, and hair follicle. In most physiologic circumstances, PTHrP carries out local rather than systemic actions. PTHrP is discussed fully in Chapter 21.
Calcitonin (CT) is a 32-amino-acid peptide whose principal function is to inhibit osteoclast-mediated bone resorption. CT is secreted by parafollicular C cells of the thyroid. These are neuroendocrine cells derived from the ultimobranchial body, which fuses with the posterior lobes of the thyroid to become the C cell mass (about 0.1% of the thyroid gland).
The secretion of CT is under the control of the ionized [Ca2+]. The C cell uses the same CaSR as the parathyroid cell to sense changes in the ambient ionized [Ca2+]. In contrast to parathyroid cells, C cells increase secretion of CT in response to hypercalcemia and shut off hormone secretion during hypocalcemia.
CT is a member of the CT gene–related peptide (CGRP) superfamily that includes: CT, CGRP, CT-receptor–stimulating peptide (CRSP), adrenomedullin, and amylin. CT and CGRP are encoded by one gene, and the respective peptides are generated from alternative splicing of primary RNA transcripts in a tissue-selective manner with CT made predominantly in the thyroid C cells. The genomic structure of the CT/CGRP/CRSP gene family is shown in Figure 8–6. In humans, there are two genes (CALCA and CALCB) that encode CGRPs and CT and one pseudogene (CALCP). Transcription of CALCA produces CT and CGRP in a tissue-specific manner by alternative splicing. The CALCB gene has a coding CGRP-beta exon and an exon that codes for CT but in which there is an in-frame stop codon. Thus, CALCB generates only CGRP-beta. In the thyroid C cell, mature CT (Figure 8–7) is incorporated within a 141-amino-acid precursor. In neurons of the central nervous system, CGRP is produced from a 128-amino-acid precursor. CGRP is a 37-amino-acid peptide with considerable homology to CT. The amino termini of both peptides incorporate a seven-member disulfide-bonded ring (see Figure 8–7). Acting through its own receptor, CGRP is among the most potent vasodilator substances known. When administered intravenously, CT produces a rapid and dramatic decline in levels of serum calcium and phosphate, primarily through actions on bone. The major effect of the hormone is to inhibit osteoclastic bone resorption. After exposure to CT, the morphology of the osteoclast changes rapidly. Within minutes, the cell withdraws its processes, shrinks in size, and retracts the ruffled border, the organelle of bone resorption, from the bone surface. Osteoclasts and cells of the proximal renal tubule express CT receptors. Like the PTH receptor, this is a G protein–coupled receptor with seven membrane-spanning regions that is coupled by Gs alpha to adenylyl cyclase and thereby to the generation of cAMP in target cells. CT also has renal effects. In the kidney, CT inhibits the reabsorption of phosphate, thus promoting renal phosphate excretion. CT also induces a mild natriuresis and increases the renal excretion of calcium. The renal effects of CT are not essential for its acute effect on serum calcium levels, which results from blockade of bone resorption.
Figure 8–6
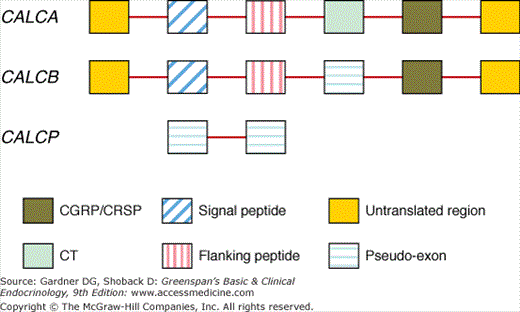
Genomic structure of the human calcitonin (CT)/calcitonin gene–related peptide (CGRP)/CT-receptor–stimulating peptide (CRSP) family of genes which includes CALCA and CALCB and the pseudogene CALCP. Alternative splicing generates the two gene products (CT in the thyroid C cells and CGRP in the nervous system) with well-defined functions in humans. Straight lines indicate the introns while the coding sequences are shown by specifically designated boxes. (Reproduced and modified, with permission from Katafuchi T, et al. Calcitonin receptor-stimulating peptide: its evolutionary and functional relationship with calcitonin/calcitonin gene-related peptide based on gene structure. Peptides. 2009;30:1753.)
Figure 8–7
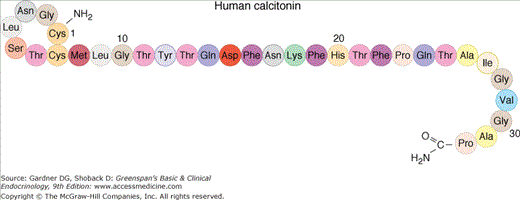
Amino acid sequence of human calcitonin, demonstrating its biochemical features, including an amino terminal disulfide bridge and carboxyl terminal prolinamide.
(Reproduced, with permission, from McPhee SJ, et al, ed. Pathophysiology of Disease: An Introduction to Clinical Medicine. Originally published by Appleton & Lange. Copyright 1995 by The McGraw-Hill Companies, Inc.)
Although its secretory control by calcium and its antiresorptive actions enable CT to counter PTH in the control of calcium homeostasis, thus engendering bihormonal regulation, it is actually unlikely that CT plays an essential physiologic role in humans and other terrestrial animals. This is supported by two lines of evidence. First, removal of the thyroid gland—the major if not the only source of CT in mammals—has no perceptible impact on calcium handling or bone metabolism in postnatal life. Second, secretion of extremely high CT levels by medullary carcinoma of the thyroid (MCT), a malignancy of the C cell, likewise has no apparent effect on mineral homeostasis. Thus, in humans, CT is a hormone in search of a function. It plays a much more obvious homeostatic role in saltwater fish, in which the major challenge is maintenance of blood calcium levels in the sea, where the ambient calcium concentration of sea water is very high.
CT is of clinical interest for two reasons. First, CT is important as a tumor marker in MCT. Second, CT has therapeutic uses as an inhibitor of osteoclastic bone resorption. CT can be administered either parenterally or as a nasal spray and is used in the treatment of Paget disease of bone, hypercalcemia, and osteoporosis.
The term vitamin D (calciferol) refers to two secosteroids: vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol) (Figure 8–8). Both are produced by photolysis from naturally occurring sterol precursors. Vitamin D2 is the principal form of vitamin D available for pharmaceutical purposes other than as dietary supplements (Table 8–2). Vitamin D3 is produced from 7-dehydrocholesterol, a precursor of cholesterol found in high concentration in the skin. Although vitamin D2 is metabolized somewhat differently than vitamin D3, the active metabolites of vitamin D2 and vitamin D3 have equivalent biologic activity so the term vitamin D, unless otherwise qualified, will be understood here to denote both. Vitamins D2 and D3 differ in their side chains: vitamin D2 has a methyl group at C24 and a double bond at C22 to C23. These features alter the metabolism of vitamin D2 compared with vitamin D3; however, both are converted to 25-hydroxyvitamin D (25[OH]D) and 1,25-dihydroxyvitamin D (1,25[OH]2D).
| Cholecalciferol, Ergocalciferol | Paricalcitol | Doxercalciferol | Calcitriol | |
|---|---|---|---|---|
| Abbreviation | D3, D2 | 19-nor calcitriol | 1αOHD2 | 1,25(OH)2D |
| Physiologic dose | 2.5-10 ug (1 ug = 40 units) | 1-5 ug | 0.25-0.5 ug | |
| Pharmacologic dose | 0.625-5 mg | 1-4 μg po2.5 μg/mL IV | 20-200 ug | 1-3 ug |
| Duration of action | 1-3 mo | Hours to days | Hours to days | Hours to days |
| Clinical applications | Vitamin D deficiency Vitamin D malabsorption Hypoparathyroidism | Secondary hyperparathyroidism of chronic renal failure | Secondary hyperparathyroidism of chronic renal failure | Secondary hyperparathyroidism of chronic renal failure Hypoparathyroidism Hypophosphatemic rickets Acute hypocalcemia Vitamin D-dependent rickets types I and II |
Figure 8–8
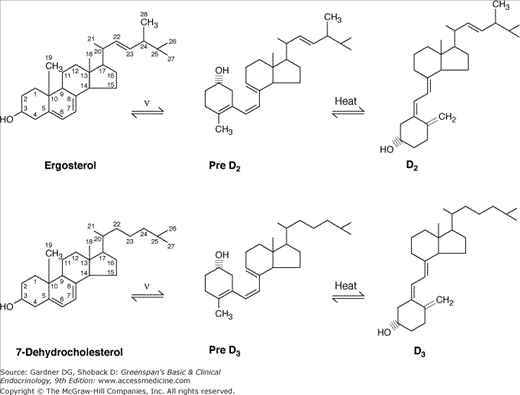
The photolysis of ergosterol and 7-dehydrocholesterol to vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol), respectively. An intermediate is formed after photolysis, which undergoes a thermal-activated isomerization to the final form of vitamin D. The rotation of the A-ring puts the 3-beta-hydroxyl group into a different orientation with respect to the plane of the A-ring during production of vitamin D.
In the process of forming vitamins D2 and D3, the B ring of the sterol precursor is cleaved, and the A ring is rotated around the C5 to C6 double bond so that the 3β-hydroxyl group is positioned below the plane of the A ring. By convention, this hydroxyl group retains its designation as 3β, and the one position above the plane of the A ring is designated 1α. Hydroxylations in the side chain can lead to stereoisomers designated R and S. The natural position for the C24 hydroxyl group is R. Both the R and the S positions can be hydroxylated at C25 in the formation of 25,26(OH)2D.
Because vitamin D can be formed in vivo (in the epidermis) in the presence of adequate amounts of ultraviolet light, it is more properly considered a hormone (or prohormone) than a vitamin. To be biologically active, vitamin D must be metabolized further. The liver is the major but not the only organ that can metabolize vitamin D to its principal circulating form, 25(OH)D. The kidney and other tissues metabolize 25(OH)D to a variety of other metabolites, the most important of which are 1,25(OH)2D and, perhaps, 24,25(OH)2D. A large number of other metabolites have been identified, but their physiologic roles are unclear. They may represent products destined only for elimination. Normal circulating levels of the principal metabolites are listed in Table 8–3. The recommended daily allowance for vitamin D in adults is 400 units (1 unit = 0.025 μg vitamin D), although recent data indicate that is inadequate especially in older individuals.
| Name | Abbreviation | Generic Name | Serum Concentrationa |
|---|---|---|---|
Vitamin D Vitamin D3 Vitamin D2 | D D3 D2 | Calciferol Cholecalciferol Ergocalciferol | 1.6 ± 0.4 ng/mL |
| 25-Hydroxyvitamin D | 25(OH)D | Calcifediol | 26.5 ± 5.3 ng/mL |
| 1,25-Dihydroxyvitamin D | 1,25(OH)2D | Calcitriol | 34.1 ± 0.4 pg/mL |
| 24,25-Dihydroxyvitamin D | 24,25(OH)2D | 1.3 ± 0.4 ng/mL | |
| 25,26-Dihydroxyvitamin D | 25,26(OH)2D | 0.5 ± 0.1 ng/mL |
Vitamin D3 is formed in the skin from 7-dehydrocholesterol, which is distributed throughout the epidermis and dermis but has its highest concentration in the lower layers of the epidermis, the stratum spinosum, and stratum basale. These epidermal layers also account for the highest production of vitamin D. The cleavage of the B ring of 7-dehydrocholesterol to form previtamin D3 (see Figure 8–8) requires ultraviolet light with a spectrum of 280 to 320 nm (UVB). Following cleavage of the B ring, the previtamin D3 undergoes thermal isomerization to vitamin D3 but also to the biologically inactive compounds lumisterol and tachysterol. Formation of pre-D3 is rapid and reaches a maximum within hours during exposure to solar or ultraviolet irradiation. The degree of epidermal pigmentation, the age of the skin, and the intensity of exposure all affect the time required to reach the maximum pre-D3 concentration but do not alter that maximum. Continued ultraviolet light exposure then results in continued formation of the inactive compounds from pre-D3. The formation of lumisterol is reversible, so lumisterol can be converted back to pre-D3 as pre-D3 levels fall. Short exposure to sunlight causes prolonged release of vitamin D3 from the exposed skin because of the slow conversion of pre-D3 to vitamin D3 and the conversion of lumisterol to pre-D3. Prolonged exposure to sunlight does not produce toxic quantities of vitamin D3 because of the photoconversion of pre-D3 to lumisterol and tachysterol.
Vitamin D3 transport from the skin into the circulation has not been thoroughly studied. Vitamin D3 is carried in the bloodstream primarily bound to vitamin D–binding protein (DBP), an α-globulin produced in the liver. DBP has a lower affinity for vitamin D3 than other vitamin D metabolites such as 25(OH)D and 24,25(OH)2D. 7-Dehydrocholesterol, pre-D3, lumisterol, and tachysterol bind to DBP even less well. Therefore, vitamin D3 could be selectively removed from skin by the gradient established by selective binding to DBP. Because the deepest levels of the epidermis make the most vitamin D3 when the skin is irradiated, the distance over which vitamin D3 must diffuse to reach the circulation is short. However, simple diffusion is an unlikely means for so hydrophobic a molecule to enter the bloodstream. Epidermal lipoproteins may play a role in transport, but this remains to be established.
Dietary sources of vitamin D are clinically important because exposure to ultraviolet light may not be sufficient to maintain adequate production of vitamin D in the skin. The farther away from the equator one lives, the shorter the period of the year during which the intensity of sunlight is sufficient to produce vitamin D3. Most milk products in the United States are supplemented with vitamin D. Unfortified milk products contain little or no vitamin D. Although plants and mushrooms contain ergosterol, their content of vitamin D2 is limited unless they are irradiated with ultraviolet light during processing. Vitamin D is found in moderate to high concentrations in fish oils and fish liver and in lesser concentrations in eggs. Vitamin D is absorbed from the diet in the small intestine with the help of bile salts. Drugs that bind bile salts such as colestipol and various malabsorption syndromes reduce vitamin D absorption. Most of the vitamin D passes into the lymph in chylomicrons, but a significant amount is absorbed directly into the portal system. The presence of fat in the lumen decreases vitamin D absorption. 25(OH)D and calcitriol are preferentially absorbed into the portal system and are less influenced by the amount of fat in the lumen. Biliary conjugates of the vitamin D metabolites have been identified, and an enterohepatic circulation of these metabolites has been established. Vitamin D is taken up rapidly by the liver and is metabolized to 25(OH)D. 25(OH)D is also transported in the blood bound to DBP. Little vitamin D is stored in the liver. Excess vitamin D is stored in adipose tissue and muscle.
As noted above, vitamin D metabolites are transported in the blood bound principally to DBP (85%) and albumin (15%). DBP binds 25(OH)D and 24,25(OH)2D with approximately 30 times greater affinity than it binds 1,25(OH)2D and vitamin D. DBP circulates at a concentration (5 × 10−6 mol/L) approximately 50 times greater than the total concentrations of the vitamin D metabolites. DBP levels are reduced in liver disease and in the nephrotic syndrome and increased during pregnancy and with estrogen administration, but are not altered by states of vitamin D deficiency or excess. The high affinity of DBP for the vitamin D metabolites and the large excess-binding capacity maintain the free and presumed biologically active concentrations of the vitamin D metabolites at very low levels—approximately 0.03% and 0.4% of the total 25(OH)D and 1,25(OH)2D levels, respectively. Liver disease reduces the total level of the vitamin D metabolites commensurate with the reduction in DBP and albumin levels, but the free concentrations of the vitamin D metabolites remain normal in most subjects. This fact must be borne in mind when evaluating a patient with liver disease to determine whether such a patient is truly vitamin D deficient. Pregnancy, on the other hand, increases both the free and total concentrations by increasing DBP levels, altering the binding of the metabolites to DBP, and increasing the production of 1,25(OH)2D. The binding of the vitamin D metabolites to DBP appears to occur at the same site, and saturation of this site by one metabolite can displace the other metabolites. This is an important consideration in vitamin D toxicity because the very high levels of 25(OH)D that characterize this condition displace the often normal levels of 1,25(OH)2D, leading to elevated free and biologically active concentrations of 1,25(OH)2D. This phenomenon at least partially explains the hypercalcemia and hypercalciuria that mark vitamin D intoxication even in patients with normal total 1,25(OH)2D levels.
At present, it is not clear whether DBP functions just to maintain a circulating reservoir of vitamin D metabolites or whether DBP participates in transporting the vitamin D metabolites to their target tissues. A different set of proteins related to heat shock proteins are found within the cell that bind the vitamin D metabolites and appear to facilitate their further metabolism. These proteins are distinct from DBP and the nuclear hormone receptors for calcitriol, which will be discussed subsequently.
The conversion of vitamin D to 25(OH)D occurs principally in the liver (Figure 8–9). Both mitochondria and microsomes have the capacity to produce 25(OH)D but with different kinetics and with different enzymes. The mitochondrial enzyme has been identified as the 5βcholestane-3α,7α,12α-triol-27(26)-hydroxylase (CYP27A1), a cytochrome P450 mixed-function oxidase, and its cDNA has been isolated. A number of microsomal P450 enzymes have been shown to have 25-hydroxylase activity with variable preference for D2 or D3 and their 1-hydroxylated metabolites. The microsomal 25-hydroxylase that has received the most attention is CYP2R1. Like the mitochondrial enzyme, these are not specific for vitamin D. Regulation of 25(OH)D production is difficult to demonstrate. Drugs such as phenytoin and phenobarbital reduce serum 25(OH)D levels, primarily by increased catabolism of 25(OH)D and vitamin D. Liver disease leads to reduced total (but not necessarily free) serum 25(OH)D levels, primarily because of reduced DBP and albumin synthesis and not reduced synthesis of 25(OH)D. Vitamin D deficiency is marked by low blood levels of 25(OH)D, primarily because of lack of substrate for the 25-hydroxylase. Vitamin D intoxication, on the other hand, leads to increased levels of 25(OH)D because of the lack of feedback inhibition on the 25-hydroxylases.
Figure 8–9
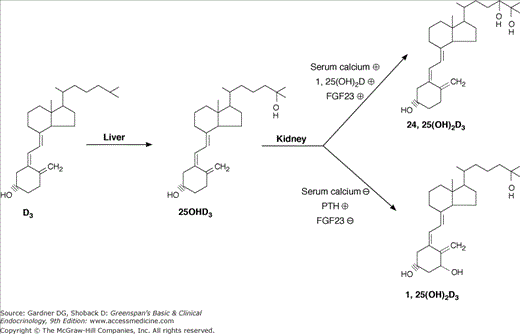
The metabolism of vitamin D. The liver converts vitamin D to 25(OH)D. The kidney converts 25(OH)D to 1,25(OH)2D and 24,25(OH)2D. Control of metabolism is exerted primarily at the level of the kidney where low serum phosphate, low serum calcium, and high parathyroid hormone (PTH) levels favor production of 1,25(OH)2D whereas FGF23; high serum calcium and phosphate, and 1,25(OH)2D inhibit 1,25(OH)2D production while increasing 24,25(OH)2D production. Plus (+) and minus (−) signs denote the stimulatory and inhibitory enzymatic reactions, respectively, driving the metabolic steps indicated.
Control of vitamin D metabolism is exerted principally in the kidney (see Figure 8–9). 1,25(OH)2D and 24,25(OH)2D are produced by cytochrome mixed-function oxidases in mitochondria of the proximal tubules. cDNAs encoding these enzymes have both been identified and show substantial homology to each other and to the mitochondrial 25-hydroxylase (CYP27A1). Their homology to other mitochondrial steroid hydroxylases is considerable but not as high. Although the 24-hydroxylase is widely distributed, the 1-hydroxylase is more limited, being found primarily in the epidermis, placenta, bone, macrophages, and prostate in addition to kidney, although low levels of expression have been observed in a number of other tissues such as breast, lung, testes, T and B lymphocytes, dendritic cells, heart, and brain.
The kidney remains the principal source of circulating 1,25(OH)2D. Production of 1,25(OH)2D in the kidney is stimulated by PTH and insulin-like growth factor-1 (IGF-1) and is inhibited by fibroblast-derived growth factor 23 (FGF23) and high blood levels of calcium and phosphate. Production of FGF23, a recently identified cytokine, occurs primarily in bone and is regulated by the levels of serum phosphate and 1,25(OH)2D (Figure 8–10). FGF23 inhibits 1,25(OH)2D production, blocks renal phosphate reabsorption, and acts to lower serum phosphate (see Figure 8–10 and Chapter 21).
Figure 8–10
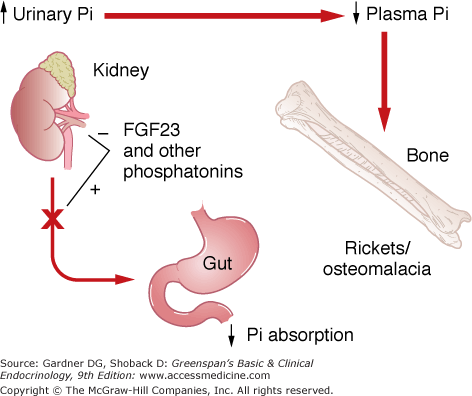
Regulation of phosphate homeostasis and 1,25(OH)2D production by FGF23 and other phosphatonins. FGF23 decreases phosphate reabsorption by the kidney and lowers plasma phosphate levels. FGF23 also blocks 1,25(OH)2D synthesis by the kidney, which reduces intestinal calcium and phosphate absorption. Excessive FGF23 production and/or action result in abnormal bone matrix mineralization, causing rickets or osteomalacia.
Calcium and phosphate also have both direct and indirect effects on 1,25(OH)2D production. In addition to direct actions on 1-hydroxylase to reduce its activity, calcium inhibits PTH secretion by the parathyroid glands, and this lowers 1,25(OH)2D production. Phosphate inhibits the production of growth hormone by the pituitary gland, leading to decreased IGF-1 generation, and stimulates FGF23 production, each step contributing to the indirect inhibition of 1,25(OH)2D production by phosphate.
1,25(OH)2D inhibits its own synthesis and stimulates the production of 24,25(OH)2D and 1,24,25(OH)3D by the 24-hydroxylase, which are the initial products of both 25(OH)D and 1,25(OH)2D catabolism, respectively. Other hormones such as prolactin and CT may regulate 1,25(OH)2D production, but their roles under normal physiologic conditions in humans have not been demonstrated.
Extrarenal production of 1,25(OH)2D is regulated in a cell-specific fashion. Cytokines such as γ-interferon (γ-IFN) and tumor necrosis factor-α (TNF-α) stimulate 1,25(OH)2D production by macrophages and keratinocytes. 1,25(OH)2D and calcium have minimal effects on 1,25(OH)2D production by these cells. This is important in understanding the pathophysiology of hypercalcemia and the increased 1,25(OH)2D levels in patients with sarcoidosis, lymphomas, and other granulomatous diseases.
The main function of vitamin D metabolites is the regulation of calcium and phosphate homeostasis, which occurs in conjunction with PTH. The gut, kidney, and bone are the principal target tissues for this regulation. The major pathologic complication of vitamin D deficiency is rickets (in children with open epiphyses) or osteomalacia (in adults), which results mainly from the deficiency of calcium and phosphate required for bone mineralization. 1,25(OH)2D is the most biologically active, if not the only vitamin D metabolite involved in maintaining calcium and phosphate homeostasis.
Most of the cellular processes regulated by 1,25(OH)2D involve the nuclear vitamin D receptor (VDR), a 50-kDa protein related by structural and functional homology to a large class of nuclear hormone receptors including the steroid hormone receptors, thyroid hormone receptors (TRs), retinoic acid and retinoid X receptors (RARs, RXRs), and endogenous metabolite receptors, including peroxisome proliferation activator receptors (PPARs), liver X receptors (LXRs), and farnesoid X receptors (FXRs) (see Chapter 1). These receptors are transcription factors. The VDR, as is the case with TRs, RARs, PPARs, LXRs, and FXRs, generally acts by forming heterodimers with RXRs. The VDR–RXR complex then binds to specific regions within the regulatory portions of the genes whose expression is controlled by 1,25(OH)2D (Figure 8–11). The regulatory regions are called vitamin D response elements (VDREs) and are generally composed of two stretches of six nucleotides (each called a half-site) with a specific and nearly identical sequence separated by three nucleotides with a less specific sequence (a direct repeat with three base-pair separation; DR3). Numerous exceptions to this rule can be found such as a VDR–RAR complex binding to a DR6 (direct repeat with six base-pair separation) or inverted palindromes (repeat elements going in opposite directions) with various degrees of spacing (eg, IP9). Other nuclear hormone receptors bind to similar elements but with different spacing or orientation of the half-sites. The binding of the VDR–RXR complex to the VDRE then attracts a number of other proteins called coactivators, which may function to open up the gene for transcription by acetylating the histones covering the gene or by bridging the gap between the VDRE and the initiation complex containing the RNA polymerase to signal the beginning of transcription. Not all actions of 1,25(OH)2D, however, can be explained by changes in gene expression. VDR in the membrane and a membrane-bound protein called the 1,25D3-membrane–associated rapid response steroid binding protein (1,25D3-MARRS) may mediate the rapid effects of 1,25(OH)2D on calcium influx and protein kinase C activity observed in a number of tissues, effects too rapid to be explained by genomic activity.
Figure 8–11
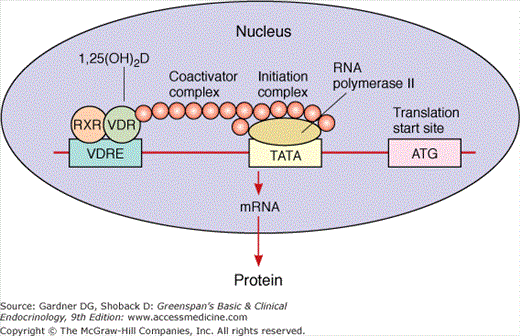
1,25(OH)2D-initiated gene transcription. 1,25(OH)2D enters the target cell and binds to its receptor, VDR. The VDR then heterodimerizes with the retinoid X receptor, RXR. This increases the affinity of the VDR–RXR complex for the vitamin D response element (VDRE), a specific sequence of nucleotides in the promoter region of a vitamin D responsive gene. Binding of the VDR–RXR complex to the VDRE attracts a complex of proteins termed coactivators to the VDR–RXR complex, some of which open up the gene for transcription by acetylating the histones and other proteins which communicate with and activate the RNA polymerase II complex. Transcription of the gene is initiated to produce the corresponding mRNA, which leaves the nucleus for the cytoplasm to be translated to the corresponding protein.
Intestinal calcium transport is the best-understood target tissue response of 1,25(OH)2D. Calcium transport through the intestinal epithelium proceeds by at least three distinct steps: (1) entrance into the cell from the lumen across the brush border membrane down a steep electrochemical gradient; (2) passage through the cytosol, probably within subcellular organelles such as mitochondria and endosomes; and (3) removal from the cell against a steep electrochemical gradient at the basolateral membrane. Each of these steps is regulated by 1,25(OH)2D. At the brush border, 1,25(OH)2D induces a change in the binding of calmodulin to brush border myosin 1, a unique form of myosin found only in the intestine, where it resides primarily in the microvillus bound to actin and to the plasma membrane. The calmodulin-myosin 1 complex may provide the mechanism for removing calcium from the brush border after it crosses the membrane into the cell. Changes in the phospholipid composition of the brush border may explain the increased flux of calcium across this membrane after calcitriol administration. None of these changes requires new protein synthesis. However, evidence now indicates that a calcium channel, the transient receptor potential cation channel 6 (TRPV6) in the brush border membrane is induced by 1,25(OH)2D and is likely the major mechanism by which calcium enters the intestinal epithelial cell. The transport of calcium through the cytosol requires a vitamin D-inducible protein called calbindin. Calbindin exists either in a 28-kDa or 9-kDa form, depending on the species and tissue. Calbindin has a high affinity for calcium. If its synthesis is blocked, the calcium content of the cytosol and mitochondria increases, and efficiency of transport is reduced. At the basolateral membrane, calcium is removed from the cell by an ATP-driven pump, the Ca2+-ATPase (PMCA1b), a protein also induced by 1,25(OH)2D. Recent studies, however, in which calbindin or TRPV6 expression is deleted show surprisingly little impact on intestinal calcium transport (calbindin knockout) or only a partial reduction (TRPV6 knockout) suggesting that there is more to learn about the mechanisms governing intestinal calcium transport. Nevertheless in mice in which the VDR is rendered nonfunctional (VDR knockout), intestinal calcium transport is markedly impaired, indicating the importance of VDR function—presumably via regulation of gene expression—in governing intestinal calcium transport.
The critical role of 1,25(OH)2D in regulating bone formation and resorption is evidenced by the development of rickets in children who are vitamin D-deficient, have inactivating mutations in CYP27B1 (the 1-hydroxylase) (pseudovitamin D-deficient rickets), or lack a functioning VDR (hereditary 1,25[OH]2D-resistant rickets). The histologic and radiologic appearance of bone in these genetic conditions is quite similar to that in patients with severe vitamin D deficiency.
Patients with inactivating mutations in the 1-hydroxylase gene can be treated successfully with calcitriol (but not with vitamin D), whereas those with mutations in the VDR are resistant to calcitriol as well as to vitamin D. These experiments of nature, however, do not exclude the possibility that other vitamin D metabolites, in combination with 1,25(OH)2D, are essential for normal bone metabolism.
In vitamin D deficiency, all vitamin D metabolites are reduced, but 25(OH)D and 24,25(OH)2D tend to be reduced out of proportion to 1,25(OH)2D, which may be maintained in the low normal range despite clear abnormalities in bone mineralization. Thus, it is reasonable to ask whether 1,25(OH)2D is the only vitamin D metabolite of consequence for the regulation of skeletal homeostasis. In fact, a number of studies point to unique actions of 24,25(OH)2D that cannot be replicated by 1,25(OH)2D, especially in cartilage, although this remains controversial. Studies in mice, in which the 24-hydroxylase gene has been inactivated (knocked out), support the concept that 24,25(OH)2D has a role in bone formation that is not replaced by 1,25(OH)2D. At least some of the skeletal abnormalities in 24-hydroxylase–null animals may be explained by their inability to regulate 1,25(OH)2D levels. Nevertheless, the need for more than just 1,25(OH)2D is an important concept in the treatment of vitamin D deficiency; this is best done with vitamin D or 25(OH)D rather than calcitriol alone, because vitamin D serves as a precursor for both 1,25(OH)2D and 24,25(OH)2D.
The mechanisms by which 1,25(OH)2D regulates skeletal homeostasis remain uncertain. Provision of adequate calcium and phosphate for mineralization is clearly important. The rickets present in patients with defective VDRs can be cured with calcium and phosphate infusions. Similarly, in mice in which the VDR is rendered nonfunctional (knocked out), rickets is prevented with diets high in calcium and phosphate. Thus, even though the osteoblast, the cell responsible for forming new bone, contains a VDR and the transcription of a number of proteins in bone is regulated by 1,25(OH)2D, it is not clear how essential the direct actions of 1,25(OH)2D on bone are for bone formation and mineralization. Thus, the bone is a good example of a tissue in which 1,25(OH)2D and calcium work together, such that limited quantities of one may be compensated for by increased amounts of the other.
In organ cultures of bone, the best established action of 1,25(OH)2D is to stimulate bone resorption. This is accompanied by an increase in osteoclast number and activity and decreased collagen synthesis. The increase in osteoclast number and activity is now known to be mediated by the production in osteoblasts of a membrane-bound protein called receptor activator of NF-kappa B ligand (RANKL), which acts on its receptor (RANK) in osteoclasts and their precursors to stimulate osteoclast differentiation and activity (see later). 1,25(OH)2D is one of several hormones (PTH and selected cytokines are others) that stimulate RANKL production. Vitamin D deficiency does not decrease bone resorption, probably because PTH levels are elevated. High levels of PTH could, therefore, compensate for the lack of 1,25(OH)2D at the level of RANKL production. The presence of partial skeletal resistance to PTH in patients with vitamin D deficiency may be explained by this mechanism.
1,25(OH)2D also promotes the differentiation of osteoblasts. This action is less well-delineated than the promotion of osteoclast differentiation. Osteoblasts pass through a well-defined sequence of biochemical processes as they differentiate from proliferating osteoprogenitor cells to cells capable of producing and mineralizing matrix. The effect of 1,25(OH)2D on the osteoblast depends on the stage of differentiation at which the 1,25(OH)2D is administered. Early in the differentiation process, in cells characterized as immature osteoblast-like cells, 1,25(OH)2D stimulates collagen production and alkaline phosphatase activity. These functions are inhibited in more mature osteoblasts. On the other hand, osteocalcin production is stimulated by 1,25(OH)2D only in mature osteoblasts. Experiments in vivo demonstrating that excessive 1,25(OH)2D can inhibit normal bone mineralization—leading to the paradoxical appearance of osteomalacia—can be understood in the context of these differential effects of 1,25(OH)2D on the osteoblast as it differentiates. Excess 1,25(OH)2D can disrupt the differentiation pathway.
The kidney expresses VDRs, and 1,25(OH)2D stimulates the expression of calbindin, TRPV5 (the renal homolog of the intestinal TRPV6), and Ca2+-ATPase (PMCA) in the distal tubule as well as 24,25(OH)2D production in the proximal tubule. However, the role of 1,25(OH)2D in regulating calcium and phosphate transport across the renal epithelium remains controversial. 25(OH)D may be more important than 1,25(OH)2D in acutely stimulating calcium and phosphate reabsorption by the kidney tubules. In vivo studies are complicated by the effect of 1,25(OH)2D on other hormones, particularly PTH, which appears to be more important than the vitamin D metabolites in regulating calcium and phosphate handling by the kidney.
An exciting observation is that VDRs are found in a large number of tissues beyond the classic target tissues—gut, bone, and kidney—and these tissues respond to 1,25(OH)2D. Furthermore, many of these tissues contain CYP27B1 (the 1-hydroxylase) and so are capable of producing their own 1,25(OH)2D from circulating 25(OH)D. These tissues include elements of the hematopoietic and immune systems; cardiac, skeletal, and smooth muscle; and brain, liver, breast, endothelium, skin (keratinocytes, melanocytes, and fibroblasts), and endocrine glands (pituitary, parathyroid, pancreatic islets [B cells], adrenal cortex and medulla, thyroid, ovary, and testis). Furthermore, malignancies developing in these tissues often contain VDRs and respond to the antiproliferative actions of 1,25(OH)2D.
The responses of these tissues to 1,25(OH)2D are as varied as the tissues themselves. 1,25(OH)2D regulates hormone production and secretion, including stimulation of insulin secretion from the pancreas and prolactin from the pituitary, but inhibiting PTH secretion from the parathyroid gland, renin secretion from the kidney, and atrial naturetic peptides from the heart. 1,25(OH)2D enables the innate immune system by inducing antimicrobial peptides such as cathelidicin but suppresses the adaptive immune response by blocking the maturation of dendritic cells and shifting the balance of T-helper cell differentiation from Th1 and Th17 to Th2 and Treg. Myocardial contractility and vascular tone are modulated by 1,25(OH)2D. 1,25(OH)2D reduces the rate of proliferation of many cell lines, including normal keratinocytes, fibroblasts, lymphocytes, and thymocytes as well as abnormal cells of mammary, skeletal, intestinal, lymphatic, and myeloid origin. Differentiation of numerous normal cell types, including keratinocytes, lymphocytes, hematopoietic cells, intestinal epithelial cells, osteoblasts, and osteoclasts as well as abnormal cells of the same lineage is enhanced by 1,25(OH)2D. Thus, the potential for manipulating a vast array of physiologic and pathologic processes with calcitriol and its analogs is enormous. This promise is starting to be realized in that vitamin D analogs are being used to treat psoriasis, uremic hyperparathyroidism, and osteoporosis. Epidemiologic and animal studies indicate a role for vitamin D in the prevention and treatment of both type 1 and type 2 diabetes mellitus, autoimmune diseases such as multiple sclerosis and inflammatory bowel disease, and infections such as tuberculosis. Trials of vitamin D analogs in the treatment of a variety of cancers are also being conducted. Thus, although regulation of bone mineral homeostasis remains the major physiologic function of vitamin D and its metabolites, clinical applications for these compounds and their analogs are being found outside the classic target tissues.
How Vitamin D and PTH Control Mineral Homeostasis
Consider a person who switches from a high normal to a low normal intake of calcium and phosphate—from 1200 mg/d to 300 mg/d of calcium (the equivalent of leaving three glasses of milk out of the daily diet). The net absorption of calcium falls sharply, causing a transient decrease in the serum calcium level. The homeostatic response to this transient hypocalcemia is led by an increase in PTH, which stimulates the release of calcium and phosphate from bone and the retention of calcium by the kidney. The phosphaturic effect of PTH allows elimination of phosphate, which is resorbed from bone together with calcium. In addition, the increase in PTH, along with the fall in serum calcium and serum phosphate, activates renal 1,25(OH)2D synthesis. In turn, 1,25(OH)2D increases the fractional absorption of calcium and further increases bone resorption. External calcium balance is thus restored by increased fractional absorption of calcium and increased bone resorption at the expense of higher steady state levels of PTH and 1,25(OH)2D.
MCT, a neoplasm of thyroidal C cells, accounts for 5% to 10% of all thyroid malignancies. Approximately 75% of MCTs are sporadic. The remainder is familial and associated with one of three heritable syndromes: familial isolated MCT; multiple endocrine neoplasia 2A (MEN 2A), consisting of MCT, pheochromocytoma, and primary hyperparathyroidism; or multiple endocrine neoplasia 2B (MEN 2B), consisting of MCT, pheochromocytoma, multiple mucosal neuromas, and, rarely, primary hyperparathyroidism (Table 8–4). The MEN syndromes are more extensively discussed in Chapter 22.
| Disorder | Characteristic Components |
|---|---|
| MEN 2A | MCT, pheochromocytoma, primary hyperparathyroidism |
| MEN 2A with lichen amyloidosis | MEN 2A + pruritic lesions of upper back |
| Familial MCT | MCT |
| MEN 2A or Familial MCT and Hirschsprung disease | MCT, Hirschsprung disease |
| MEN 2B | MCT, pheochromocytoma, marfanoid habitus, intestinal and mucosal ganglioneuromatosis |
Understanding the pathogenesis of MCT has been greatly enhanced by the identification of causative mutations in the RET proto-oncogene located on chromosome 10q11.2. The RET gene encodes a membrane tyrosine kinase receptor whose ligands belong to the glial cell line-derived neurotrophic factor (GDNF) family. This receptor is expressed developmentally in migrating neural crest cells that give rise to hormone-secreting neuroendocrine cells (eg, C cells and adrenal medullary cells) and to the parasympathetic and sympathetic ganglia of the peripheral nervous system. Remarkably, different mutations in RET can produce five distinct diseases (see Table 8–4). Inheritance of certain activating mutations is responsible for MEN 2A and familial MCT. Inheritance of a different set of activating mutations causes MEN 2B. In over half of sporadic MCTs, the tumor has a clonal somatic mutation (present in the tumor but not in genomic DNA), which is identical to one of the mutations that is responsible for the familial forms of MCT. These somatic mutations clearly play a causative role in sporadic MCT. Mutations in the RET gene can also produce Hirschsprung disease, a congenital absence of the enteric parasympathetic ganglia, that can lead to a disturbance in intestinal motility, resulting in megacolon.
MCT is usually located in the middle or upper portions of the thyroid lobes. It is typically unilateral in sporadic cases but often multicentric and bilateral in familial forms of the disease. The natural history of MCT is variable. Sporadic tumors may be quite aggressive or very indolent; the mean 5-year survival rate is about 50%. The behavior of familial forms varies among syndromes. MEN 2B has the most aggressive form of MCT, with a 2-year survival of about 50%; MEN 2A has a course similar to that of sporadic forms of this tumor, and familial MCT has the most indolent course of all MCT. The natural history of the tumor in familial MCT, MEN 2A, and MEN 2B syndromes has been dramatically impacted by early prophylactic total thyroidectomy that is now advised in most carriers of disease-causing RET mutations. MCT may spread to regional lymph nodes or undergo hematogenous spread to the lungs and other viscera. When metastatic, this tumor is sometimes associated with a chronic diarrhea syndrome. The pathogenesis of the diarrhea is unclear. In addition to CT, these tumors secrete a variety of other bioactive products, including prostaglandins, serotonin, histamine, and peptide hormones (ACTH, somatostatin, corticotropin-releasing hormone). In some cases, the associated diarrhea responds dramatically to treatment with long-acting somatostatin analog such as octreotide, which blocks secretion of these bioactive products.
CT is a tumor marker for MCT. It is most sensitive for this purpose when secretion is stimulated with provocative agents. The standard provocative tests use pentagastrin (0.5 ug/kg intravenously over 5 seconds) or a rapid infusion of calcium gluconate (2 mg calcium/kg over 1 minute). Blood samples are obtained at baseline and 1, 2, and 5 minutes after the stimulus. For maximal sensitivity, both tests are usually combined, with the calcium infusion immediately followed by administration of pentagastrin. Although basal CT levels are often normal in early tumors, CT levels may be many times higher than normal in patients with disseminated MCT. Despite this, the patients are uniformly normocalcemic. Although tumors secrete larger molecular weight forms of CT with decreased biologic activity, monomeric CT levels are often high as well. RET oncogene analysis has replaced provocative testing in most cases, although basal CT levels are still used to follow disease burden postoperatively and during chemotherapy trials.
Members of families carrying RET mutations must be screened for MCT and, if positive, for the associated tumors that occur in MEN 2A and MEN 2B (see Table 8–4 and Chapter 22). In the case of MCT, the presence of the RET mutation in an individual generally leads to the recommendation for a total thyroidectomy prior to the development of frank malignancy or abnormal CT levels. It is recommended in a kindred with a known RET mutation that children be screened at birth for the carrier state. Total thyroidectomy with or without central compartment lymph node dissection can be performed in mutation carriers, and further testing can be discontinued in genetically normal family members. The timing of prophylactic thyroidectomy in carriers of RET mutations, however, depends on the genotype present (ie, codon mutated in RET) (Table 8–5). As supported by the genotype-phenotype correlations in these diseases, surgery is recommended in certain high-risk cases before 1 year of age, in cases of intermediate risk before 5 years of age, and in lower-risk patients later in childhood usually by 10 years of age. Because a limited number of mutations in RET cause more than 95% of hereditary MCT and up to 25% of sporadic disease, it is possible to screen the majority of patients using commercial reference laboratories.
| Codon | Frequency (Percent of Total) |
|---|---|
| MEN 2Aa | |
| 634 | 87 |
| 620 | 6 |
| 618 | 3 |
| 611 | 2 |
| 609 | <1 |
| MEN 2Bb | |
| 918 | 94 |
| 883 | 5 |
| 804 | <1 |
| Familial MCTc | |
| 618 | 30 |
| 634 | 26 |
| 620 | 21 |
| 768 | 8 |
| 609 | 4 |
| 804 | 3 |
| 790, 791, 891 | Uncertain |
Apparent sporadic MCT also calls for genetic testing and family studies. Up to 25% of new cases of MCT may actually be probands of families who harbor one of the familial syndromes. It has been noted in some kindreds after careful and lengthy follow-up that some individuals develop pheochromocytomas or primary hyperparathyroidism. Therefore, patients with apparent familial MCT should be followed indefinitely for one of these components of MEN 2A. Screening of family members can be accomplished by genetic testing, once the index case is identified. Identification of a mutation that is present only in tumor tissue would establish the mutation as somatic and the tumor as a sporadic one. Identification of the same RET mutation in tumor and genomic DNA would make the diagnosis of a familial form of the disorder and would mandate careful screening of the family.
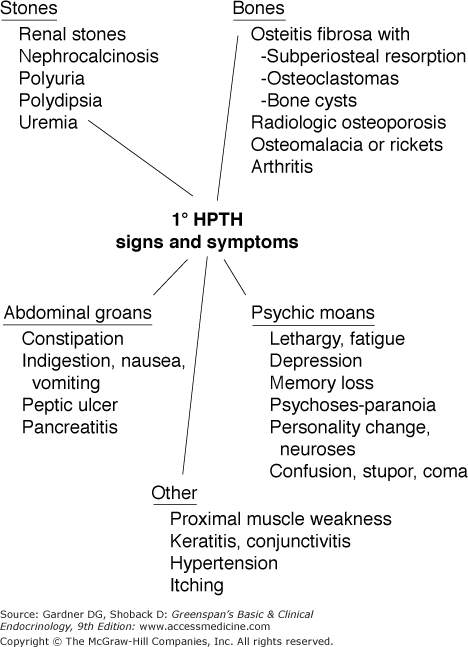
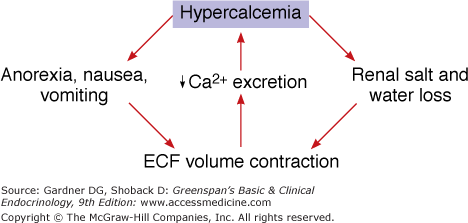
A number of symptoms and signs accompany hypercalcemia. They include central nervous system effects such as lethargy, depression, psychosis, ataxia, stupor, and coma; neuromuscular effects such as weakness, proximal myopathy, and hypertonia; cardiovascular effects such as hypertension, bradycardia (and eventually asystole), and a shortened QT interval; renal effects such as stones, decreased glomerular filtration, polyuria, hyperchloremic acidosis, and nephrocalcinosis; gastrointestinal effects such as nausea, vomiting, constipation, and anorexia; eye findings such as band keratopathy; and systemic metastatic calcification. Primary hyperparathyroidism, one of the most common etiologies for hypercalcemia, has the mnemonic for recalling its signs and symptoms as stones, bones, abdominal groans, and psychic moans (Figure 8–12).
Although many disorders are associated with hypercalcemia (Table 8–6), they produce it by a limited number of mechanisms: (1) increased bone resorption, (2) increased gastrointestinal absorption of calcium, or (3) decreased renal excretion of calcium. Although any of these mechanisms can be involved in a given patient, the common feature of virtually all hypercalcemic disorders is accelerated bone resorption. The only hypercalcemic disorder in which bone resorption does not play a part is the milk-alkali syndrome.
Primary hyperparathyroidism Sporadic Associated with MEN 1 or MEN 2A Familial Postrenal transplantation |
Variant forms of hyperparathyroidism Familial benign hypocalciuric hypercalcemia Lithium therapy Tertiary hyperparathyroidism in chronic renal failure |
Malignancies Humoral hypercalcemia of malignancy Caused by PTHrP (solid tumors, adult T-cell leukemia syndrome) Caused by 1,25(OH)2D (lymphomas) Caused by ectopic secretion of PTH (rare) Local osteolytic hypercalcemia (multiple myeloma, leukemia, lymphoma) |
| Sarcoidosis or other granulomatous diseases |
Endocrinopathies Thyrotoxicosis Adrenal insufficiency Pheochromocytoma VI Poma |
Drug induced Vitamin A intoxication Vitamin D intoxication Thiazide diuretics Lithium Milk-alkali syndrome Estrogens, androgens, tamoxifen (in breast carcinoma) |
| Immobilization |
| Acute renal failure |
| Idiopathic hypercalcemia of infancy |
| ICU hypercalcemia |
| Serum protein disorders |
The central feature of the defense against hypercalcemia is suppression of PTH secretion. This reduces bone resorption, renal production of 1,25(OH)2D, and, thereby, intestinal calcium absorption and increases urinary calcium losses. The kidney plays a key role in the adaptive response to hypercalcemia as the only route of net calcium elimination. The level of renal calcium excretion is markedly increased by the combined effects of an increased filtered load of calcium and the suppression of PTH. However, the patient who relies on the kidneys to excrete an increased calcium load is in precarious balance. Glomerular filtration is impaired by hypercalcemia; the urinary concentrating ability is diminished, predisposing to dehydration; poor mentation may interfere with access to fluids; and nausea and vomiting may further predispose to dehydration and renal azotemia. Renal insufficiency, in turn, compromises calcium clearance, leading to a downward spiral (Figure 8–13). Thus, once established, many hypercalcemic states are self-perpetuating or aggravated through the vicious cycle of hypercalcemia. The only alternative to the renal route for elimination of calcium from the extracellular fluid is deposition of calcium phosphate and other salts in bone and soft tissues. Soft tissue calcification is observed with massive calcium loads, with massive phosphate loads (as in tumor lysis syndrome, crush injuries and compartment syndromes), and when renal function is markedly impaired and the calcium-phosphate product rises.
As a practical matter, the categories for differential diagnosis are primary hyperparathyroidism and everything else (see Table 8–6). Hyperparathyroidism is by far the most common cause of hypercalcemia and has distinctive pathophysiologic features. Thus, the first step in the differential diagnosis is determination of intact PTH (Figure 8–14). If the PTH level is high, and thus inappropriate for hypercalcemia, little further workup is required except to consider the variant forms of hyperparathyroidism that are discussed later. If the PTH level is suppressed, then a search for other entities must be conducted. Most other entities in Table 8–6 are readily diagnosed by their distinctive features, as discussed later.
Primary hyperparathyroidism results from the excessive secretion of PTH and typically produces frank hypercalcemia. With the advent of multiphasic screening of serum chemistries, we have come to recognize that primary hyperparathyroidism is a common and usually asymptomatic disorder. Its incidence is approximately 42 per 100,000, and its prevalence is up to 4 per 1000 in women over age 60. Primary hyperparathyroidism is approximately 2 to 3 times more common in women than in men.
Primary hyperparathyroidism is caused by a single parathyroid adenoma in about 80% of cases and by primary hyperplasia of the parathyroids in 10% to 15%. Parathyroid carcinoma is a rare cause of hyperparathyroidism, accounting for 1% to 2% of cases. Parathyroid carcinoma is often recognizable preoperatively because it presents with severe hypercalcemia or a palpable neck mass.
Sporadic parathyroid adenomas have a clonal origin, indicating that they can be traced back to an oncogenic mutation in a single progenitor cell. A few of these genetic alterations have been identified and/or assigned to chromosomal loci. About 25% of sporadic parathyroid adenomas have chromosomal deletions involving chromosome 11q12-13 that are thought to eliminate the putative tumor suppressor gene MENIN. As reviewed later and in Chapter 22, loss of the functioning menin protein, a tumor suppressor, is the cause of parathyroid, pituitary, and pancreatic tumors in the MEN 1 syndrome. An additional 40% of parathyroid adenomas display allelic loss on chromosome 1p (1p32pter).
Another important locus on chromosome 11 that has been implicated in approximately 4% of sporadic parathyroid adenomas involves the cyclin D1 gene. In the initial elucidation of this pathogenic mechanism, a chromosomal rearrangement was found in a parathyroid tumor. Breakage and then inversion of a piece of chromosome 11 led to the expression of cyclin D1, a cell-cycle regulatory protein, under the control of the PTH promoter. As expected, this promoter is highly active in parathyroid cells, resulting in marked overexpression of cyclin D1 in these tumor cells. Cyclin D1 is normally expressed at high levels in the G1 phase of the cell cycle and permits entry of cells into the mitotic phase of the cycle. Thus, a parathyroid-specific disorder of cell-cycle regulation leads to abnormal cell proliferation and ultimately excessive PTH production.
Parathyroid hyperplasia accounts for 12% to 15% of cases of primary hyperparathyroidism and is the pathologic basis for these inherited conditions: MEN 1, MEN 2A, the hyperparathyroidism-jaw tumor (HPT-JT) syndromes, and isolated familial hyperparathyroidism. In all of these instances four or fewer glands may be involved at the time of presentation. Inheritance of all of these conditions is autosomal dominant.
Parathyroid hyperplasia was traditionally viewed as an example of true hyperplasia, a polyclonal expansion of cell mass. This occurs in other endocrine tissues when a trophic hormone present in excess (eg, ACTH excess) induces bilateral adrenal hyperplasia. Molecular analysis, however, has revised this view. Parathyroid hyperplasia appears to share similar pathogenic mechanisms with parathyroid adenomas. The MEN 1 syndrome illustrates this. MEN 1 is due to the inherited inactivation of one allele of the MENIN gene, which encodes a tumor suppressor. Acquired postnatal somatic mutations in the remaining MENIN gene result in loss of the other allele’s function. This leads to tumors in those endocrine tissues in which the gene is expressed. In this view, multicentric somatic mutations would account for the occurrence of four-gland hyperparathyroidism.
Kindreds with a presentation like MEN 1 but without mutations in MENIN have been investigated for germline mutations in cyclin-dependent kinase inhibitors (CDKIs) of which there are seven known genes. In an analysis of 196 consecutive cases of MEN 1 or other tumor states without germline mutations in MENIN, one laboratory found seven probable pathologic mutations in these genes. Three of the seven mutations were in p27 with the remainder in three other CDKIs (p15, p18, and p27). These mutations are thought to be disease causing because abnormalities in CDKI–protein interaction were uncovered in the majority of these mutations tested. The CDKIs negatively control the cell cycle through interaction with and inhibition of cyclins D, E, and A. The germline mutations in these kindreds would, therefore, involve loss of function in known inhibitors of cell growth and cell cycle activity.
In MEN 2A, the occurrence of parathyroid hyperplasia is also a consequence of expression of activating mutations of the RET gene in the four glands. These and other studies clearly indicate that parathyroid hyperplasia is typically monoclonal in its origin, implying that cell hyperproliferation arose from a single progenitor cell in each gland.
In addition to hyperparathyroidism, as one part of classical MEN 1, it is now clear that a subset of cases of familial isolated hyperparathyroidism is due to germline mutations in the MENIN gene. This observation established the concept that isolated familial hyperparathyroidism can be an allelic variant of the MEN 1 syndrome. The variation from MEN 1 lies in the absence of tumors in the other endocrine glands.
Other cases of familial isolated hyperparathyroidism are explained, albeit rarely, by mutations in the gene that causes the HPT-JT syndrome (see later). Additional families with isolated hyperparathyroidism have, as yet, no identified mutations.
In contrast to MEN 1 and MEN 2A, both benign and malignant parathyroid tumors occur in the HPT-JT syndrome. This is an autosomal dominant disorder that includes primary hyperparathyroidism (90%), jaw tumors (ossifying fibromas of the mandible or maxilla) (30%), renal cysts (10%), and less frequently renal hamartomas and Wilms tumor. Linkage analysis originally determined the locus for this syndrome, which had been designated as hyperparathyroidism 2 or HRPT2 to be at 1q24-q32. Inactivating mutations in HRPT2 gene, encoding the protein parafibromin, have been identified in HPT-JT kindreds. Mutations in the HRPT2 gene inactivate parafibromin. This protein like menin normally functions as a tumor suppressor. Its function(s) in parathyroid cells have not yet been elucidated.
Inactivating mutations in the HRPT2 gene have also been identified in kindreds with isolated familial hyperparathyroidism. Both single- and multiple-gland disease have been noted. It was further noted that approximately 15% of affected individuals with HPT-JT syndrome develop parathyroid cancer, instead of adenomas or hyperplasia, suggesting that reduced parafibromin function also contributes to a malignant phenotype. Several groups have shown that a large percentage (~70%) of patients with sporadic parathyroid cancer harbor somatic mutations in the HRPT2 gene. Surprisingly, approximately 30% of individuals with isolated parathyroid cancer have also been found to have germline mutations in HRPT2 without the other manifestations of the HPT-JT syndrome. It is now thought that the majority of sporadically occurring parathyroid cancers are due to somatic mutations in the coding region of the HRPT2 gene. It is anticipated that mutations in the noncoding regulatory regions of this gene may explain additional cases. A pathologic study of more than 50 cases of parathyroid cancer indicates that the loss of parafibromin immunoreactivity is a specific and key feature of malignancy with a sensitivity of 96% (CI, 85%-99%) and specificity of 99% (CI, 92%-100%).
The typical clinical presentation of primary hyperparathyroidism has evolved considerably over the past few decades. As the disease continues to be detected primarily by multiphasic screening that includes determination of serum calcium levels, there has been a marked reduction in the frequency of the classic signs and symptoms of primary hyperparathyroidism. Renal disease (stones, decreased renal function, and occasionally nephrocalcinosis) and the classic hyperparathyroid bone disease osteitis fibrosa cystica are decidedly rare today. In fact, about 85% of patients presenting today have neither bone nor renal manifestations of hyperparathyroidism and are regarded as asymptomatic or at most minimally symptomatic. At the same time, we have begun to recognize more subtle manifestations of hyperparathyroidism. This has presented a number of questions about the role of parathyroid surgery in primary hyperparathyroidism, which are discussed later (see Treatment).
Hyperparathyroid bone disease—The classic bone disease of hyperparathyroidism is osteitis fibrosa cystica. Formerly common, this disorder now occurs in less than 10% of patients. Clinically, osteitis fibrosa cystica causes bone pain and sometimes pathologic fractures. The most common laboratory finding is an elevation of the alkaline phosphatase level, reflecting high bone turnover. Histologically, there is an increase in the number of bone-resorbing osteoclasts, marrow fibrosis, and cystic lesions that may contain fibrous tissue (brown tumors). The most sensitive and specific radiologic finding of osteitis fibrosa cystica is subperiosteal resorption of cortical bone, best seen in high-resolution films of the phalanges (Figure 8–15A). A similar process in the skull leads to a salt-and-pepper appearance (Figure 8–15B). Bone cysts or brown tumors may present as osteolytic lesions. Dental films may disclose loss of the lamina dura of the teeth, but this is a nonspecific finding also seen in periodontal disease.
The other important skeletal consequence of hyperparathyroidism is osteoporosis. Unlike other osteoporotic disorders, hyperparathyroidism often results in the preferential loss of cortical bone (Figure 8–16). In general, both the mass and mechanical strength of trabecular bone are relatively maintained in mild primary hyperparathyroidism. Patients who are followed medically with this disease generally do not experience progressive bone loss for as long as 8 years after diagnosis (Figure 8–17). This may be due to the fact that mild PTH excess has an anabolic effect on the skeleton to maintain or even increase bone mass. Although osteoporosis, defined by bone mineral density (BMD) T-scores by dual-energy x-ray absorptiometry (DXA) of −2.5 or lower, is generally considered to be an indication for surgical treatment of primary hyperparathyroidism, the impact of low bone mass on morbidity is difficult to corroborate clinically (see Treatment of Hypercalcemia).
Hyperparathyroid kidney disease—Once common in primary hyperparathyroidism, kidney stones now occur in 10% to 20% of cases depending on the series. These are usually calcium oxalate stones. From the perspective of a stone clinic, only about 7% of calcium stone formers prove to have primary hyperparathyroidism. They are difficult to manage medically, and stones constitute one of the agreed indications for parathyroidectomy. Clinically evident nephrocalcinosis rarely occurs, but a gradual loss of renal function is not uncommon. Renal function is stabilized after a successful parathyroidectomy, and otherwise unexplained renal insufficiency (defined as an estimated glomerular filtration rate [eGFR] <60 mL/min) in the setting of primary hyperparathyroidism is also considered to be an indication for surgery because of the risk of progression. Chronic hypercalcemia can also compromise the renal concentrating ability, giving rise to polydipsia and polyuria.
Nonspecific features of primary hyperparathyroidism—Although stupor and coma occur in severe hypercalcemia, the degree to which milder impairments of central nervous system function affect the typical patient with primary hyperparathyroidism is unclear. Lethargy, fatigue, depression, difficulty in concentrating, and personality changes may occur; however, some patients appear to benefit from parathyroidectomy. Frank psychosis also responds to surgery on occasion. Muscle weakness with characteristic electromyographic changes is also seen, and there is evidence from controlled clinical trials that surgery can improve muscle strength. A number of studies have examined quality of life (QOL) and psychological function in patients with primary hyperparathyroidism pre- and post-parathyroid surgery, compared to a medically followed group. Results vary as to whether QOL improves with surgery. It was formerly thought that the incidence of hypertension was increased in primary hyperparathyroidism, but more recent evidence suggests that it is probably no greater than in age-matched controls, and parathyroidectomy appears to be of no benefit. Dyspepsia, nausea, and constipation all occur, probably as a consequence of hypercalcemia, but there is probably no increase in the incidence of peptic ulcer disease. The articular manifestations of primary hyperparathyroidism include chondrocalcinosis in up to 5% of patients. Acute attacks of pseudogout, however, are less frequent.
Figure 8–15
A: Magnified x-ray of index finger on fine-grain industrial film showing classic subperiosteal resorption in a patient with severe primary hyperparathyroidism. Note the left (radial) surface of the distal phalanx, where the cortex is almost completely resorbed, leaving only fine wisps of cortical bone. B: Skull x-ray from a patient with severe secondary hyperparathyroidism due to end-stage renal disease. Extensive areas of demineralization alternate with areas of increased bone density, resulting in the “salt and pepper” skull x-ray. (Both films courtesy of Dr. Harry Genant.)
Figure 8–16
Related posts:
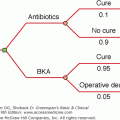


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


