Fig. 2.1
Preprinted orders for potassium replacement
The oral route is preferred over other routes if feasible.
The intravenous route may be used in patients with profound hypokalemia or unable to tolerate oral replacement. The rate of intravenous administration should not exceed 20 mEq/h diluted in intravenous fluid through a peripheral vein. The infusion rate may be as high as 40 mEq/h through a central venous catheter.
In general, the relationship between the degree of hypokalemia and total body deficit is linear. For each 1-mEq/L decrease in serum potassium level, the total body deficit would be about 300 mEq. This total body deficit may be corrected over days.
About 40–50 % of patients with hypokalemia also have hypomagnesemia, which must be corrected to fully correct the potassium-depleted state.
Potassium-sparing diuretics, such as amiloride and spironolactone, inhibit potassium excretion and may have a role in decreasing renal potassium wasting.
Hyperkalemia
Hyperkalemia is also a common electrolyte disorder in cancer patients.
Clinical Manifestations
Severe clinical manifestations of hyperkalemia usually are absent until the serum potassium level is greater than 7.5 mEq/L. Some patients (e.g., those with chronic renal failure) can tolerate high serum potassium levels without having any clinical signs or symptoms. At greater than 7.5 mEq/L, nonspecific symp toms such as muscle weakness, cramping, and paralysis of different muscle groups may occur.
Hyperkalemia causes depolarization of excitable membranes. This membrane depolarization leads to the excitability of nerves and muscles, causing cramps, muscle weakness, and paralysis. The most vital organ with excitable membranes is the heart. Specific electrocardiogram (EKG) changes and potentially fatal arrhythmias may be present, but the serum potassium level is not correlated directly with a particular EKG pattern. An early EKG abnormality associated with hyperkalemia is peak T waves followed by a progressive QRS widening to a “sinusoidal” wave. Ventricular tachycardia, fibrillation, and asystole may occur.
Approach
Inappropriate potassium content in intravenous fluid and total parenteral nutrition are com mon iatrogenic causes of hyperkalemia. A significant release of intracellular potassium will cause hyperkalemia, as in the case of tumor ly sis syndrome. Insulin deficiency, β-blocker therapy, and acidemia can elevate serum potassium levels.
Drug-induced hyperkalemia most often occurs in patients with impaired renal excretion of potassium. The drugs commonly used by cancer patients that may cause hyperkalemia include cyclosporin A, tacrolimus, heparin, mitomycin C, and pentamidine.
Diminished renal excretion of potassium occurs in patients with acute or chronic renal failure, renal hypoperfusion, or type 4 renal tubular acidosis. Drugs that can lead to decreased potassium excretion include potassium-sparing diuretics and angiotensin-converting enzyme inhibitors.
Treatment
Treatment of hyperkalemia depends on its severity and rate of development.
If possible, discontinue medications that may contribute to hyperkalemia, such as β-adrenergic blockers, nonsteroidal anti-inflammatory drugs, angiotensin-converting enzyme inhibitors, potassium supplements, and others described above.
Monitor EKG continuously if the potassium level is greater than 6 mEq/L.
For EKG changes, infuse intravenously (usually for less than 60 min):
Calcium (1–2 g of calcium gluconate or 0.5–1.0 g of chloride)
Sodium bicarbonate
Glucose (usually 25 g) plus 6–8 U of regular insulin
β-adrenergic agonists, which promote potassium entry into cells
Increasing the renal excretion of potassium can be attempted with the use of loop diuretics.
Removal of potassium from the body should be attempted with the use of ion exchange resins, such as sodium polystyrene sulfonate (Kayexalate ), which can be administered orally (15–30 g/dose) or rectally (30–60 g/dose) as a retention enema.
Emergent hemodialysis may be used in refractory cases.
Hypocalcemia
In hospitalized cancer patients, the hypocalcemia rate is about 13.4 %. Hypocalcemia may affect the proper functioning of many intracellular and extracellular processes, such as muscle contraction, nerve conduction, and blood coagulation.
Clinical Manifestations
Hypocalcemia can be asymptomatic if it is mild. Life-t hreatening problems such as seizures, cardiac dysrhythmias, and laryngospasm can occur if hypocalcemia is severe. Acute hypocalcemia is characterized by neuromuscular irritability. Acute symptoms are mus cle weakness, paresthesia, spasm, tetany, hyperreflexia, Chvostek sign, Trousseau sign, seizure, bronchospasm, laryngeal spasm, and respiratory failure. Cardiovascular presentations are bradycardia, hypotension, QT-interval prolongation, congestive heart failure, and cardiac arrest. Chronic hypocalcemia with hypoparathyroidism causes extrapyramidal disorders, cataracts, and skin and hair changes. Vitamin D deficiency causes rickets and osteomalacia in patients with hypocalcemia.
Approach
In most cancer patients, the etiology of hypocalcemia is obvious. Excluding a decreased serum calcium level owing to low albumin and serum protein levels, the major causes of hypocalcemia are hypoparathyroidism and hypomagnesemia. Hypocalcemia may be a feature of tumor lysis syndrome. Severe osteoblastic bone metastases (especially from prostate carcinoma) are often associated with hypocalcemia. The toxicity of certain chemotherapeutic agents (e.g., platinum compounds) also may lead to hypocalcemia.
Evaluation of hypocalcemia involves confirmation of it by measuring the ionized calcium level. If the cause of hypocalcemia is not clear, laboratory analysis of intact parathyroid hormone (PTH) , magnesium, phosphate, 25-hydroxy vitamin D3, 1,25-dihydroxy vitamin D3, creatinine, and 24-hour urinary calcium levels is helpful.
Treatment
Treatment of hypocalcemia depends on its severity and cause.
Severe hypocalcemia is treated parenterally with intravenous calcium chloride (0.5–1.0 g) or gluconate (1–2 g) over 5–10 min. Calcium gluconate is preferred over other agents because it is less likely to cause tissue necrosis if extravasated.
Hypomagnesemia is a common cause of hypocalcemia. Concurrent hypomagnesemia should be treated with intravenous magnesium sulfate followed by oral replacement.
Chronic hypocalcemia is treated with oral calcium preparations (e.g., gluconate, carbonate) containing 1–2 g of elemental calcium per day. Patients with hypoparathyroidism often must receive lifelong supplementation of calcium and vitamin D. Vitamin D supplements can be given in 1-hydroxylated form or as calcitriol. Calcitriol is preferred for patients with renal insufficiency or failure because of decreased 1-α-hydroxylase levels in the kidneys.
Attention should be paid to phosphate binding.
Hypercalcemia
Hypercalcemia of malignancy is observed in 10–15 % of cancer patients. It is a poor prognostic sign that is associated with short survival durations.
Clinical Manifestation
Patients with mild hypercalcemia (calcium level less than 12 mg/dL) usually have no symptoms, whereas those with moderate or severe hypercalcemia are frequently symptomatic. Central nervo us system symptoms are lethargy, ataxia, stupor, coma, mental status changes, and psychosis. Gastrointestinal tract symptoms are anorexia, nausea, constipation, ileus, dyspepsia, and pancreatitis. Renal signs are polyuria, nephrolithiasis, and nephrocalcinosis. Cardiovascular manifestations can be a short QT interval, ST segment depression, sinus arrest, and atrioventricular block. Musculoskeletal symptoms are myalgia, arthralgia, and weakness.
Approach
Hypercalcemia may result from increased bone resorption, renal tubular reabsorption, and gastrointestinal absorption of calcium. In cancer patients, hypercalcemia of malignancy accounts for more than 90 % of hypercalcemia cases. Hypercalcemia in cancer patients may have different pathophysiologic mechanisms. The most common humor al factor secreted by tumors causing hypercalcemia is PTH-related peptide. In general, patients with PTH-related peptide-induced hypercalcemia have advanced malignant disease and poor prognoses. Other humoral factors, such as interleukin-1 and -6, prostaglandins, and tumor necrosis factor, can mediate hypercalcemia in cancer patients. Extensive lytic bone metastasis, particularly in patients with breast cancer or multiple myeloma, may lead to hypercalcemia. Increased levels of 1,25-dihydroxy vitamin D3 may mediate hypercalcemia in patients with Hodgkin disease or non-Hodgkin lymphoma.
Serum calcium levels should be interpreted in the con text of protein binding (corrected calcium level = [0.8 × (normal albumin level − patient’s albumin level)] + serum calcium level). However, accurate measurement of the ionized calcium level confirms hypercalcemia. Laboratory studies of the following help diagnose the etiology of hypercalcemia: intact PTH, PTH-related protein, 25-hydroxy vitamin D3, and 1,25-dihydroxy vitamin D3.
Treatment
Treatment of hypercalcemia should be aimed at lowering seru m calcium levels and correcting its underlying causes, if possible.
Primary hyperparathyroidism can be cured via parathyroidectomy .
Use of medications (e.g., calcium-containing medications, thiazide diuretics) that contribute to hypercalcemia should be discontinued.
The initial and first-line treatment of hypercalcemia is hydration with crystalloid intravenous fluid. In patients with overall fluid overload, use of a loop diuretic would be helpful.
Bisphosphonates (etidronate, clodronate, pamidronate, zoledronate, and ibandronate) inhibit bone resorption by osteoclasts. Zoledronate (4–6 mg intravenously over 30 min) is more widely used than pamidronate (60–90 mg intravenously over 4–24 h).
Second-line agents include calcitonin (salmon calcitonin 4 IU/kg subcutaneously every 12 h). Calcitonin has a rapid onset of action, although its effectiveness may decrease within 2–3 days.
Other less widely used agents include glucocorticoids, plicamycin (25 μg/kg intravenously), and gallium nitrate (200 mg/m2 intravenously).
Hypomagnesemia
The incidence rate of the common electrolyte deficiency hypomagnesemia in hospitalized cancer patients has been as high as 17.1 %. Hypomagnesemia is defined as a plasma serum concentration of magnesium less than 1.5 mg/dL. However, magnesium levels that are persistently less than 1.8 mg/dL would indicate depletion of total body magnesium.
Clinical Manifestations
Magnesium is a major cation in the body, and only 1–2 % of total body magnesium is present in the extracellular space. It is needed for a wide variety of enzymatic reactions, including those involving ATP and nucleic acid metabolism. Magnesium is also directly involved in the regulation of calcium and potassium metabolism. The clinical manifestations of hypomagnesemia may be nonspecific and include anorexia, nausea, vomiting, lethargy, dizziness, muscle weakness, tremor, muscle fasciculation, tetany, and tonic-clonic seizures.
Approach
In cancer patients, hypomagnesemia is a very common abn ormality that is related t o low intake and impairment of renal reabsorption or intestinal absorption of magnesium. It is also related to prolonged intravenous feeding, nasogastric suction, chronic alcoholism, intestinal malabsorption, and diarrhea. The renal toxicity of chemotherapy (e.g., platinum-based drugs, cyclophosphamide, ifosfamide) and anti-infective medications (e.g., amphotericin, aminoglycosides) also influences hypomagnesemia.
Hypomagnesemia is often associated with other electrolyte abnormalities, such as hypokalemia and hypocalcemia. Concurrent measurement of other electrolytes, such as calcium, phosphate, and potassium, should be considered.
Treatment
Magnesium replacement is indicated in cancer patients when the serum magnesium level is repeatedly below normal (Fig. 2.2).
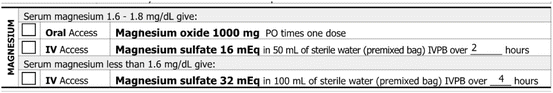
Fig. 2.2
Preprinted orders for magnesium replacement
1.
Oral replacement is preferred over parenteral when feasible. However, diarrhea may be a dose-limiting side effect.
2.
When intravenous replacement is required, the usual practice is to replace half of the estimated dose over 1 day and the remaining half over the next 3–4 days.
Hypermagnesemia
Hypermagnesemia is uncommon. It is usually caused by increased intake of magnesium in the presence of renal insufficiency or iatroge nic factors.
Clinical Manifestations
The clinical manifestations of hypermagnesemia correlate well with the serum level of magnesium. Early signs include nausea, vomiting, weakness, and cutaneous flushing, which can occur when the serum magnesium level is greater than 3 mg/dL. With levels greater than 4 mg/dL, hyporeflexia and loss of deep tendon reflexes may occur. At levels greater than 5 mg/dL, hypotension and EKG changes (QRS widening, QT and PR prolongation, and conduction abnormalities) may occur. Respiratory depression, coma, and complete heart block may occur at levels greater than 9 mg/dL. Asystole and cardiac arrest can occur at levels greater than 10 mg/dL.
Approach
The major causes of hypermagnesemia are renal failure and excessive ingestion of magnesium-containing medications in the presence of renal insufficiency. In the absence of renal insufficiency, hypermagnesemia owing to excessive intake of magnesium is very rare, as excess magnesium in the gastrointestinal tract leads to diarrhea. Overreplacement of magnesium in intravenous fluid or with hyperalimentation also can cause hypermagnesemia. A less common cause in cancer patients is tumor lysis syndrome.
Excessive magnesium intake usually is evident in the patient’s dietary and medication history. Hypermagnesemia is diagnosed via direct measurement of serum magnesium levels. Renal function should be assessed by measuring blood urea nitrogen and creatinine levels.
Treatment
Discontinuation of magnesium intake is the first step.
Patients with mild symptoms and normal renal function can simply be observed to ensure that the magnesium level returns to normal.
Magnesium excretion can be accelerated by hydration with crystalloid fluid and a loop diuretic given intravenously.
In cases of severe hypermagnesemia (particularly with hypotension and/or cardiac arrhythmia), calcium should be administered intravenously to reverse respiratory depression, hypotension, and cardiac arrhythmia.
Emergent dialysis should be considered for patients with life-threateni ng hypermagnesemia.
Hypophosphatemia
Hypophosphatemia is quite prevalent, as it is found in about 2–3 % of all hospitalized patients and about 30 % of cancer patients.
Clinical Manifestations
Acute severe hypophosphatemia may lead to generalized neur ologic findings such as lethargy, confusion, disorientation, and hallucinations and focal neurologic findings such as dysarthria, dysphagia, oculomotor palsy, anisocoria, nystagmus, ataxia, cerebell ar tremor, ballismus, hyporeflexia, distal sensory deficits, paresthesia, and hyperesthesia. Severe neurologic symptoms, such as muscle paralysis, seizure, and coma, are observed only when the serum phosphate level is less than 0.8 mg/dL. Cardiac muscle also can be affected by severe hypophosphatemia, and reversible left ventricular dysfunction can occur.
Muscle weakness is the most common complaint. B one pain is another prominent complaint of phosphate-depleted patients. Prolonged hypophosphatemia leads to rickets. Hypophosphatemic rickets can result from ifosfamide nephrotoxicity. Osteomalacia, waddling gait, bone tenderness, pseudofractures, and fractures can occur in patients with chronic hypophosphatemia.
Approach
Acute hypophosphatemia occurs primarily in hospitalized patients with serious illnesses and pre-existing phosphate depletion. Acute severe hypophosphatemia usually results from translocation of phosphate into cells. Respiratory alkalosis, intravenous glucose administration (including hyperalimentation), gram-negative sepsis, and insulin therapy can induce transcellular shift of phosphate.
Chronic hypophosphatemia results from an elevated PTH or PTH-related protein level, consumption of oral phosphate binders, accelerated bone formation, increased humoral factors suppressing renal reabsorpt ion of phosphate, or intrinsic renal tubular defect in phosphate reabsorption.
Tumor-induced (oncogenic) osteomalacia is a rare syndrome characterized by hypophosphatemia, excessive urinary p hosphate loss, reduced 1,25-dihydroxy vitamin D concentrations, and osteomalacia. Tumor secretion of fibroblast growth factor 23 may be responsible for renal phosphate wasting.
Rapid cancer or normal cell proliferation in ill patients with nutritional deprivation or catabolism may cause hypophosphatemia. Chronic hypophosphatemia together with hypocalcemia occasionally is associated with extensive osteo blastic metastasis of prostate, breast, lung, and other malignancies. Patients with rapidly progressing leukemia or lymphoma (e.g., Burkitt lymphoma) may have hypophosphatemia. As with the use of granulocyte colony-stimulating factors, hematopoietic reconstitution after stem cell transplantation or stem cell harvesting in preparation for transplantation also cause hypophosphatemia.
The liver plays a significant role in phosphate homeostasis. In a retrospective study, postoperative serum phosphate levels dropped in all 44 patients who underwent right or extended right hepatic lobectomy. Authors have reported hypophosphatemia in a patient with hepatocellular carcinoma complicating liver cirrhosis. Hypophosphatemia in malnourished patients (especially alcoholics) results from a combination of magnesium deficiency, vitamin D deficiency, and malabsorption. Refeeding of high-calorie diets in severely malnourished patients can lead to refeeding syndrome with hypophosphatemia.
Intrinsic renal tubular defects in phosphate reabsorption may occur in patients with Fanconi syndrome, myeloma, or amyloidosis. Hypophosphatemia also may be associated with the use of chemotherapeutic drugs such as platinum compounds and alkylating agents (e.g., ifosfamide).
Hypophosphatemia is demonstrated via measurement of the serum phosphate level. Measurement of renal function and potassium, magnesium, calcium, vitamin D metabolite, and PTH levels is helpful in determining the cause of hypophosphatemia. If urinary loss of phosphate is suspected, urine should be collected to measure the renal phosphate threshold/glomerular filtration rate to confirm phosphaturia.
Treatment
Significant hypophosphatemia (phosphate level less than 2 mg/dL), especially in the context of underlying phosphate depletion, should be corrected promptly (Fig. 2.3).

Fig. 2.3
Preprinted orders for phosphate replacement
Phosphate can be safely administered intravenously at an initial dose of 0.2–0.8 mmol/kg over 6 h (i.e., 10–50 mmol over 6 h). Higher doses (1.5–3.0 mmol/kg over 12 h) should be reserved for patients with phosphate levels less than 1.5 mg/dL and normal renal function.
Mild hypophosphatemia can be treated with oral phosp hate in divided doses of 750–2000 mg/day.
In patients with oncogenic osteomalacia, complete resection of the tumor will reverse all biochemical abnormalities. If cure is not possible, reversal of 1,25-dihydroxy vitamin D deficiency via calcitriol administration and correction of hypophosphatemia are effective palliative therapies.
Hyperphosphatemia
Hyperphosphatemia is found in 2.5 % of cancer patients.
Clinical Manifes tations
The clinical manifestations of acute hyperphosphatemia are similar to those of associated hypocalcemia. Paresthesia, muscle cramps, tetany, and QT-interval prolongation may be induced directly by severe hyperphosphatemia. Chronic hyperphosphatemia, especially associated with hypercalcemia, may lead to diffuse visceral deposition of calcium phosphate. Deposition of calcium phosphate in the kidneys may lead to renal failure.
Approach
In the absence of renal failure, the fasting serum phosphate level is determined primarily according to the renal tubular reabsorption rate. A massive amount of phosphate can be released into the extracellular fluid via extensive cellular breakdown. Extensive rhabdomyolysis and hemolysis may cause hyperphosphatemia in the same way.
Translocation of phosphate from cells in response to metabolic or respiratory alkalosis can lead to acute hyperphosphatemia. Chronic hyperphosphatemia is present in patients with hypoparathyroidism. Excess phosphate intake (including use of phosphate-containing laxatives) is another potential cause of hyperphosphatemia.
In patients with hyperglobulinemia , pseudohyperphosphatemia must be excluded with a specimen that is free of protein (removed via precipitation with sulfosalicylic acid). In those with hyperphosphatemia, renal function must be assessed. In addition, measurement of lactic dehydrogenase, uric acid, potassium, and calcium levels is necessary in the detection and management of hyperphosphatemia owing to cellular breakdown.
Treatment
The emergency treatment of hyperphosphatemia involves supportive care and treatment of symptomatic hypocalcemia.
In patients with normal renal function, infusion of isotonic saline increases phosphate excretion.
Administration of dextrose and insulin drives phosphate into cells, temporarily lowering the serum phosphate level.
When hyperphosphatemia is life-thr eatening, hemodialysis or peritoneal analysis should be considered.
Dietary restriction of phosphorus, although an important factor in the control of the serum phosphorus level in the chronic setting, poses practical problems that limit its success in most patients.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree