Chapter Outline
Overview of Folate and Cobalamin Deficiencies
Megaloblastic Anemia of Folate and Cobalamin Deficiencies
Nonhematologic Manifestations of Cobalamin Deficiency
ACQUIRED CAUSES OF COBALAMIN DEFICIENCY
DIAGNOSTIC APPROACH TO ACQUIRED CAUSES OF COBALAMIN DEFICIENCY
ACQUIRED CAUSES OF FOLATE DEFICIENCY
DIAGNOSTIC APPROACH TO ACQUIRED CAUSES OF FOLATE DEFICIENCY
TREATMENT OF ACQUIRED DEFICIENCIES
Reversal of Megaloblastic Anemia and Other Clinical Manifestations
Management of Acquired Cobalamin Deficiencies
GENETIC DISORDERS CAUSING MEGALOBLASTIC ANEMIA
Inborn Errors of Folate Transport and Metabolism
Inborn Errors of Cobalamin Transport and Metabolism
Thiamine-Responsive Megaloblastic Anemia
The metabolism of folate and that of cobalamin are intimately intertwined, and deficiency of either vitamin can manifest as megaloblastic anemia. The two deficiencies must be rigorously differentiated and treated specifically because their causes, mechanisms, nonhematologic expressions, courses, and prognoses differ. This is as true in pediatric medicine as in adult and geriatric medicine, from which much of the existing hematologic information originated. Recent advances in molecular biology, genetics, and epigenetics have increased our knowledge of cobalamin and folate metabolism. In addition, as biochemical tools to diagnose deficiencies have become more accurate and precise, the way that we define deficiency has expanded to include biochemically defined subclinical deficiency. Epidemiologic advances have connected disorders not traditionally linked to vitamin deficiency, such as neural tube defects (NTDs) and cardiovascular disease, suggesting potential means of preventing their development. Even if some of the epidemiologic information has still unsettled implications for clinical practice and public health, the wealth of new data deserves attention. In this chapter we review the relevant clinical and scientific knowledge from all these streams, emphasize its applications in pediatric hematology, and specify wherever possible the known distinctions between infants, older children, and adults.
Definitions
Megaloblastic anemia is a specific, relatively small subset of macrocytic anemia. It is defined by a characteristic megaloblastic appearance of nucleated cells that suggests maturational nuclear-cytoplasmic asynchrony. Despite the name “anemia,” all three hematopoietic cell lines are affected noticeably as the deficiency advances. Pancytopenia can ultimately become extreme. The term pernicious anemia, sometimes misapplied to the megaloblastic anemia in cobalamin deficiency, is used here in its preferred, specific meaning: the absence of gastric intrinsic factor (IF) that produces cobalamin malabsorption and, ultimately, deficiency.
Deficiency is an elusive term with surprisingly limited conceptual and pragmatic consensus on its definition(s). Where relevant, in this chapter we will use clinical deficiency of cobalamin or folate to denote the presence of vitamin-specific clinical signs and symptoms that require medical intervention. However, most cobalamin and folate deficiencies identified today are subclinical. They are clinically silent and expressed only by (often minimal) biochemical manifestations; most importantly, the likelihood for progression is usually uncertain, and consensus does not yet exist for medical intervention or for according them the same weight as clinical deficiency.
The term cobalamin refers to all corrins with coenzyme activity in humans. Vitamin B 12 originated as the name for synthetic cyanocobalamin but is widely used as a generic synonym for cobalamin. The term folate refers to synthetic folic acid and to the various natural folate coenzymes, which are reduced dihydrofolates and tetrahydrofolates and their single-carbon substituted forms. Most folates in nature are reduced and substituted folate polyglutamates.
The term newborn is used in this chapter for a child in the first month of life and the term infant for a child in the first 2 years of life.
History
The story of megaloblastic anemia began with Addison’s description in 1849 of a puzzling disease that he believed had an adrenal origin and whose lethality Biermer immortalized by calling it “pernicious anemia.” A series of clinical and pathologic descriptions from the 1870s through the 1920s identified the hematologic and neurologic features of the disease, setting the stage for one of the landmark clinical investigations in hematology. Using reticulocyte counts as evidence, Minot and Murphy reversed the anemia by feeding patients large amounts of liver in 1926, and for their findings they were awarded a Nobel Prize. Castle then proved with elegant clinical experiments that the disease was caused by the loss of an “intrinsic factor” in gastric juice that allowed the internalization of an extrinsic factor. In the 1940s, the “extrinsic factor” was crystallized, identified, and synthesized as cyanocobalamin (vitamin B 12 ), which could then be injected intramuscularly. Readers are referred to very readable accounts of the compelling history of pernicious anemia.
Another important story was developing on a separate track, in which somewhat similar clinical syndromes involving megaloblastic anemia (“pernicious anemia of pregnancy” and “tropical anemia”) were described and shown by Wills to respond to yeast extract. Folic acid was synthesized in 1948 and shown to reverse folate deficiency. A notable self-experiment in a single subject proved that folate deficiency induced by dietary restriction produced megaloblastic anemia and mapped the evolution of the hematologic changes prospectively. After the synthesis of the two vitamins, work by many investigators solidified accurate biochemical testing for cobalamin and folate status and delineated the clinical differences between folate and cobalamin deficiencies. New tests with improved sensitivity and accuracy have been developed, although a gold standard for the diagnosis of deficiency remains elusive, especially for cobalamin deficiency.
Equally important have been the continuing advances over decades in delineating the complex transport system of each vitamin and the modern molecular and genetic understanding of the proteins and receptors involved. These advances are especially encouraging and important to pediatric care of cobalamin deficiency, which is not as common and had not heretofore been studied as intensively as in adults. The major advances in the past two decades and the explosion in diagnostic, biochemical, genetic, and therapeutic advances in identifying and treating new genetic disorders and the pathways they illuminate are reviewed within this chapter.
Folate
Chemistry of Folates
Folates are heterocyclic compounds consisting of a pteroyl group conjugated with one or more glutamic acid units ( Fig. 10-1 ). Folic acid, or pteroylglutamic acid, is an oxidized folate consisting of pteroic acid conjugated with a single glutamate residue. Biologically active folates are derived from folic acid by reduction of its pterin ring to form 7,8-dihydrofolate (DHF) and 5,6,7,8-tetrahydrofolate (THF); by addition of single carbon units at different redox states to nitrogen 5 or 10 of the pterin ring; and by conjugation of up to seven total glutamate residues by γ-carboxyl bonding. Intracellular folates exist primarily as one-carbon group substituted tetrahydrofolate polyglutamates.
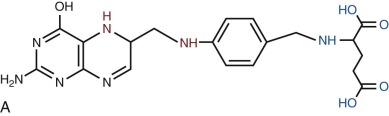
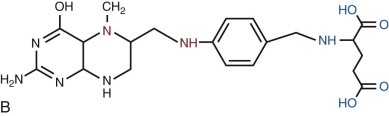
Folate derivatives that play an important role in cellular metabolism include 5-methylTHF, 5,10-methyleneTHF, 5,10-methenylTHF, 10-formylTHF, 5-formylTHF, and 5-formiminoTHF. Folic acid itself is available commercially as a stable yellow powder. It has low solubility in aqueous solutions and at acid pH. 5-FormylTHF (folinic acid, leucovorin) is a stable reduced folate that is available commercially. Other reduced folates are photolabile and susceptible to destruction by oxidation, readily breaking at the carbon 9—nitrogen 10 bond to produce inactive catabolites.
Folate Polyglutamates
Most intracellular folates exist as polyglutamylated derivatives, containing up to six or seven glutamate residues attached by γ-peptide bonds. Synthesis of these folate polyglutamates depends on activity of the enzyme folylpoly-γ-glutamate synthetase (EC 6.3.2.17), which adds glutamate residues one at a time to reduced folates, using adenosine triphosphate (ATP) as an energy source. Folate polyglutamates have higher affinity for most folate-dependent enzymes than the corresponding monoglutamates and are efficiently channeled between active sites in multifunctional enzymes. They do not readily cross the cell or mitochondrial membranes, and polyglutamylation of folates within cells thus contributes to the ability of cells to retain reduced folates in the presence of low extracellular concentrations. Separation of cytosolic and mitochondrial folate polyglutamate pools has important consequences for cellular folate metabolism.
Folate Sources, Requirements, and Stores
Folate is synthesized de novo by bacteria, plants, fungi, and certain protists. Animals are dependent on their diets for adequate supplies of the vitamin. Foods with very high folate content include dark green vegetables, orange juice, nuts, legumes, and liver. However, the reduced folates present in food are labile to light and oxidation and are partly destroyed during processing and cooking. The recommended daily allowance for folate is expressed in dietary folate equivalents to take into account the greater bioavailability of folic acid than the reduced polyglutamylated forms present naturally in food. Values recommended by the Institute of Medicine of the United States and the World Health Organization are listed in Table 10-1 .
| Age/Condition | Institute of Medicine (1998) µg/day (DFE) | World Health Organization (2004) µg/day (DFE) |
|---|---|---|
| 0-6 mo | 65 * | 80 |
| 7-12 mo | 80 * | 80 |
| 1-3 yr | 150 | 160 |
| 4-8 yr | 200 | 4-6 yr: 200 7-9 yr: 300 |
| 9-13 yr | 300 | 300 |
| >14 yr | 400 | 400 |
| Pregnancy | 600 | 600 |
| Lactation | 500 | 500 |
* Values are expressed as “adequate intake” rather than recommended daily allowance.
The average content of body stores in adults has been estimated to be 12 to 28 mg folate, based on a content of 6 to 14 mg (or 4.5 to 10 µg/g) in liver, the major storage organ. It is relevant to note here that the ratio of body folate stores to recommended daily intakes is less than 100 : 1, whereas the ratio for cobalamin approximates 1000 : 1. As a result, the folate depletion necessary to produce megaloblastic anemia is achieved much more rapidly than depletion of cobalamin. Storage data are sparse in children, but infants of normal birth weight who died in the perinatal period had hepatic folate contents of 0.76 mg ; low-birth-weight infants’ hepatic content averaged only 0.13 mg. These data, although not fully representative, suggest much lower stores–to–recommended intake ratios in newborns and infants than in adults.
Recognition that supplementation in the periconceptional period with folic acid results in decreased occurrence and recurrence of NTDs in children of women with no clinical evidence of folate deficiency led to recommendations that all women of childbearing years should take folate supplements (200 to 400 µg daily), because the critical period for prevention of NTDs is early in pregnancy before most women are aware that they are pregnant. Since January 1998, folic acid fortification of all enriched cereal-grain products has been mandated in the United States, Canada, and parts of South America. As a result, serum and red blood cell (RBC) folate concentrations have increased in these populations and the incidence of NTD-affected pregnancies has decreased by 26% in the United States and by 48% in Canada. Concerns about folate fortification delaying the diagnosis of cobalamin deficiency, promotion of cancer growth, and issues of free choice have delayed mandating folate addition to the diet in Europe and elsewhere.
The median serum folate level of 5.5 µg/L in the total National Health and Nutrition Examination Survey (NHANES) study population before fortification increased to 11.9 µg after fortification ; in children, folate level increased from 8.8 to 15.6 µg/L for ages 4 to 11 years and from 5.1 to 11.0 µg/L for ages 12 to 19 years. The mildly lower folate status in adolescents, albeit still within the reference range, is unexplained. The median RBC folate level increased from 174 to 256 µg/L in the total population, and in children it increased from 200 to 256 µg/L at ages 4 to 11 years and from 150 to 224 µg/L at 12 to 19 years. NHANES data after fortification estimated that daily folate intake averaged 626 µg in men and 534 µg in women. Intakes in children averaged 425 µg at ages 1 to 3 years, 642 µg at 4 to 8 years, 600 µg at 9 to 13 years, and 606 µg at 14 to 18 years ; the total homocysteine (tHcy) levels in the age groups between 4 and 18 years old were within the expected reference range (4.6 to 5.2 µmol/L).
Physiology of Folates
Cellular Folate Uptake
Three distinct systems for cellular uptake of folates have been identified in mammals. The reduced folate carrier (RFC) supports a low-affinity, high-capacity system for uptake of reduced folates and methotrexate (an antifolate drug) at high concentrations (in the micromolar range) with a neutral optimum pH; it is less effective in uptake of folic acid. It appears to be an important folate transporter in many types of cells, including hematopoietic cells. RFC is encoded by the SLC19A1 gene on chromosome 21q22.3. Inactivation of Slc19a1 in mice results in neonatal lethality owing to failure of erythropoiesis.
A second, high-affinity low-capacity system for folate uptake, active at nanomolar folate concentrations, is mediated by members of the folate receptor family. Several different receptors have been identified, encoded by genes on the long arm of chromosome 11 ( FOLR1, FOLR2, FOLR3, and FOLR4 ), with different forms expressed at different times in different tissues. Folate receptor-α and folate receptor-β are attached to the cell surface by a glycosylphosphatidylinositol anchor. The circulating folate-binding protein that carries some of the circulating folate is derived from folate receptor-α by cleavage from this anchor. Binding of folates to folate receptors initiates carrier-mediated endocytosis. The level of cell surface folate receptors increases greatly in cultured mouse cells grown in very low folate concentrations, suggesting that these receptors play an important role in cellular folate uptake under these conditions.
The third folate transport protein, the proton-coupled folate transporter (PCFT), has an acidic optimal pH, and transports reduced and oxidized folates with similar efficiency. It is expressed at high levels in liver, kidney, and small intestine, with lower expression in other tissues. It was originally identified as a low-affinity heme transport protein but was subsequently shown to have higher affinity for folates. PCFT is encoded by the SLC46A1 gene on chromosome 17q11.2.
Absorption of Folates from the Gut
Dietary folates are principally folate polyglutamates. These are hydrolyzed within the gut lumen or brush border, or in the lysosomes of enterocytes, by the enzyme γ-glutamylhydrolase (EC 3.4.22.12), to generate folate monoglutamates that cross cell membranes much more easily than the corresponding polyglutamates. Adequate absorption is impossible without this hydrolysis. Uptake of folate monoglutamates occurs primarily in the proximal small intestine and is mediated by PCFT. RFC is also expressed in enterocytes, but its role in this tissue remains unknown. The efficiency of absorption of folic acid, folate polyglutamates, and food folate varies in different subjects. In general, absorption of synthetic folic acid is about twice as efficient as that of folate in food whereas folic acid from folic acid–fortified cereal grain food is absorbed about 1.7 times as efficiently as food folate. The carrier-mediated process in the proximal small intestine for reduced folates is saturable, with an upper limit approximating 200 µg of food folate. The absorption of unreduced folic acid, when taken in large doses, occurs more distally by nonsaturable high-capacity mechanisms, which helps explain both the higher bioavailability of folic acid than food folates and the frequent response to oral folic acid by patients with malabsorptive disease.
Folate appears in the portal venous blood within 15 minutes of entering the stomach and reaches a maximum level in portal venous plasma about 1 hour later. Reduced folates, such as 5-formylTHF, are rapidly converted to 5-methylTHF during absorption. A second potential source of folate is that synthesized by bacteria in the colon, some of which exists as monoglutamates. There is evidence for a folate transport system, with the same properties as the small intestine system, in the colon. This may contribute to whole-body folate homeostasis, or at least support folate requirements of colon cells.
It has been estimated that up to 100 µg of folates, including folate delivered to the liver and some hepatic folate, is excreted into the bile and subsequently reabsorbed. This enterohepatic recirculation mediates a slower and more even absorption than would occur in its absence. There is evidence that enterohepatic recirculation of folates is impaired with alcohol ingestion, contributing to folate deficiency.
Transport of Folates in Plasma
Plasma folate typically consists almost entirely of 5-methylTHF, but significant amounts of folic acid may be present in individuals who have consumed folic acid supplements or food supplemented with folic acid. Most folate circulates unbound, although some may be bound to albumin and a small proportion is bound to a folate-binding protein derived from the cell surface protein folate receptor-α. Plasma folate-binding protein levels are increased in pregnancy, in folate deficiency, in renal failure, with breast and ovarian cancers, and with some types of inflammation. Its function in plasma is unclear. It is present in milk, where it may function to improve the absorption of folate by the newborn.
Folate Transport Across the Blood-Brain Barrier
The concentration of folate (primarily 5-methylTHF) in the cerebrospinal fluid (CSF) is twofold to threefold higher than in blood, indicating active transport occurs across the blood-brain barrier. Both PCFT and folate receptor-α are expressed at the choroid plexus, and mutations at either SLC46A1 or FOLR1 are associated with decreased cerebral 5-methylTHF levels. It has been suggested that folate receptor-α mediates endocytosis of folates and that PCFT is required for export of endocytosed folates from acidified endosomes.
Folate in the Kidney
Folate, free and bound to folate-binding protein or albumin, is filtered at the glomerulus and reabsorbed in the proximal tubule, with the result that little folate appears in the urine. Both folate receptor-α and folate receptor-β are expressed in the brush border of the proximal tubule and are responsible for reabsorption of free folate. Reabsorption of folate bound to albumin is carried out by the large multiligand endocytic receptors megalin and cubilin, whereas folate-binding protein is reabsorbed by megalin.
Folate Transport in the Placenta
Levels of plasma folate are higher in cord than maternal blood. Perfusion of the human placental cotyledon revealed a two-step maternal-to-fetal transfer of methylTHF, with binding of folate to placental folate-binding proteins on the maternally facing chorionic surface and gradual release generating an intervillous blood folate level three times that in the maternal circulation, followed by passive transfer to the fetal circulation along a downhill concentration gradient. There is expression of folate receptor-α, PCFT, and RFC in placenta, with folate receptor-α and PCFT localizing together to the syncytiotrophoblast microvillous plasma membrane, where they may function together in the same way that they do in the choroid plexus.
Biochemistry of Folates
Intracellular folate metabolism involves the transfer of one-carbon units derived from breakdown of a variety of metabolites. A scheme of cellular folate metabolism is illustrated in Figure 10-2 . Metabolism can be divided into reactions involved in transfer of one-carbon units to THF, generating one-carbon substituted reduced folates; interconversion of one-carbon substituted THF derivatives; and utilization of one-carbon units in amino acid and nucleic acid metabolism.
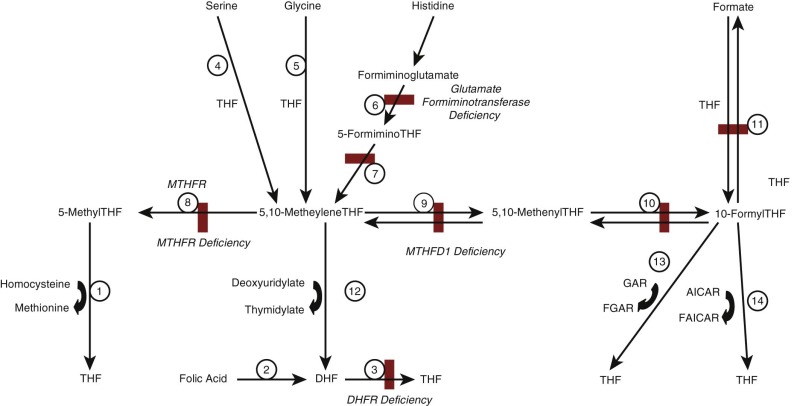
The primary source of one-carbon units for folate metabolism is catabolism of serine to form glycine, which generates 5,10-methyleneTHF (reaction 4 in Fig. 10-2 ). Additional sources include breakdown of glycine (reaction 5), dimethylglycine and sarcosine, which also generate 5,10-methyleneTHF, and breakdown of histidine, which generates 5,10-methenylTHF (reactions 6 and 7). ATP-dependent condensation of formate with THF to form 10-formylTHF is catalyzed by methylTHF synthetase (reaction 11). Once one-carbon units have been attached, the various forms can be interconverted to ensure that the cell has the folate derivatives required for metabolism. 5,10-MethyleneTHF can be reduced to form 5-methylTHF by the enzyme methylenetetrahydrofolate reductase (MTHFR; reaction 8). Alternatively, it can be oxidized to form 5,10-methyleneTHF and 10-formylTHF by the multifunctional enzymes encoded by MTHFD1 in the cytoplasm or MTHFD2/MTHFD2L in mitochondria (reactions 9 and 10).
One-carbon substituted forms of cobalamin are required for several critical steps in metabolism.
- 1.
Methylation of homocysteine to form methionine is catalyzed by methionine synthase and utilizes 5-methylTHF as methyl group donor (see Fig. 10-2 , reaction 1); this reaction also requires cobalamin and S-adenomethionine to function and represents the point in cellular metabolism where folate and cobalamin metabolism interact. Methionine is not only required for protein synthesis; it can also be converted to S-adenosylmethionine, which is the methyl group donor in a large number of transmethylation reactions that are involved in DNA and RNA methylation and in neurotransmitter synthesis, among other processes.
- 2.
Conversion of deoxyuridylate to deoxythymidylate, the final step in thymidine synthesis, utilizes 5,10-methyleneTHF as its one-carbon unit donor (see Fig. 10-2 , reaction 12). This reaction generates DHF, which must be reduced to THF before it can be used again as a one-carbon unit recipient. This reduction is accomplished by DHF reductase (see Fig. 10-2 , reaction 3).
- 3.
Two separate steps in de novo purine biosynthesis use 10-formylTHF as a one-carbon donor, generating carbons 2 and 8 of the purine ring structure (see Fig. 10-2 , reactions 13 and 14).
Folates and Arteriosclerotic Vascular Disease
Homocysteine was first implicated as an atherogenic factor by the observation of premature arteriosclerotic disease in children with severe hyperhomocysteinemia due to inborn errors of metabolism. It was subsequently suggested that moderate elevations of tHcy were associated with development of atherosclerotic disease in adults. A common thermolabile variant of MTHFR, c.677C→T (p.A222V), results in an approximately 50% decrease in enzyme activity when present in the homozygous state and in elevated tHcy in the presence of low folate concentration. Many observational studies showed an association between elevated tHcy and cardiovascular disease and a modest, but statistically significant, increased risk for cardiovascular disease in patients with the T allele. However, suggestions that lowering tHcy might improve cardiovascular outcomes in patients have not been supported in several large epidemiologic studies.
Folates and Neural Tube Defects and Other Birth Defects
As noted previously, folate supplementation during the periconceptional period (from initiation of pregnancy to closure of the neural tube at about 1 month of gestation) reduces the occurrence and recurrence of NTDs, leading to fortification of flour and other enriched grain products with folic acid in the United States and other countries. This has been associated with a decrease in prevalence of NTDs. Studies have also suggested a role for folate supplementation in preventing nonsyndromic cleft lip with or without cleft palate and congenital heart defects, but the evidence for these associations is not as strong.
Folates and Cancer
Folate status appears to have an inverse relationship with the risk for cancers of various types. However, epidemiologic studies have also suggested an association between higher folate status and progression of premalignant lesions in several tissues, and increased folate concentrations have been associated with growth of some malignant cells. The effects of folate have been most thoroughly investigated in colorectal cancer, where results of meta-analysis were consistent with a small protective effect of folate against risk for cancer. There is less convincing evidence for a protective role for folate against cancers of the oropharynx, esophagus, stomach, pancreas, lungs, cervix, ovary, and breast as well as for neuroblastoma and leukemia. A transient increase in the rate of colorectal cancer in the United States and Canada following the start of folic acid fortification of grains may reflect the effect of increased folate concentration on proliferation of existing adenomas, however. Meta-analyses of studies of folic acid supplementation (at levels tenfold higher than those used in folic acid fortification of grain) showed no effect on the rate of cancer.
Folate Antimetabolites
Antifolates are analogues of folic acid that block folate metabolism, interrupting DNA synthesis and causing cell death. Methotrexate, a potent inhibitor of mammalian DHF reductase, is effective in combination chemotherapy of carcinomas of the breast, lung, bone, ovary, head and neck, and elsewhere and contributes to cure of choriocarcinoma and childhood acute lymphoblastic leukemia. It is also used for its immunosuppressive and antiproliferative properties in the treatment of psoriasis, rheumatoid arthritis, and graft-versus-host disease after bone marrow transplantation. Newer antifolates have been synthesized and tested over the past 40 years. Some are discussed in Chapter 46 , “Chemotherapy in the Pediatric Cancer Patient.”
Pyrimethamine is an antimalarial drug with greater affinity for parasitic than human DHF reductase. Trimethoprim is an antifolate with antibacterial activity and a higher affinity for bacterial than human DHF reductase. In combination with sulfisoxazole, which interferes with bacterial synthesis of folic acid, there is a double synergistic antifolate attack on bacteria. Such combinations are often used to treat urinary tract and other infections and as prophylaxis against pneumonia resulting from infection with Pneumocystis jirovecii (formerly Pneumocystis carinii ) in immunocompromised patients.
Cobalamin (Vitamin B 12 )
Chemistry of Cobalamins
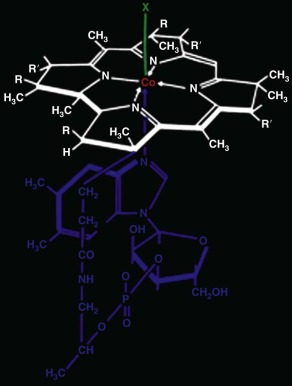
Cobalamins, members of the class of molecules called corrins, have the chemical structure shown in Figure 10-3 . They consist of a planar corrin ring, similar to but distinct from that of porphyrins such as heme, with a central cobalt atom that coordinates with four nitrogen atoms of the corrin ring. A nucleotide, 5,6-dimethylbenzimidazole, coordinates with the cobalt atom below the plane of the corrin ring and attaches covalently as well to the corrin ring. The central cobalt atom may exist in three different oxidation states: the oxidized trivalent state (cob[III]alamin), the divalent state (cob[II]alamin), and the fully reduced monovalent state (cob[I]alamin). Several different groups may be coordinated to the cobalt atom in the upper axial position (position “X” in Fig. 10-3 ) when it is in the cob(III)alamin state. Corrins that have coenzyme activity or can be converted to coenzyme forms within the cell include cobalamins with cyanide (cyanocobalamin [CNCbl]), the hydroxyl radical (hydroxocobalamin [OHCbl]), the methyl radical (methylcobalamin [MeCbl]), or 5′-deoxyadenosyl (adenosylcobalamin [AdoCbl]) bound in the upper axial position, as well as cob(II)alamin and cob(I)alamin. Under certain conditions, for example at acidic pH or when bound to certain proteins, cobalamin can exist in a “base off” conformation, in which the dimethylbenzimidazole base no longer coordinates with the central cobalt atom.
Cobalamin Sources, Requirements, and Stores
Nutritional Sources and Requirements
Although prokaryotes, animals, and many algae require cobalamin for metabolism, only prokaryotes synthesize it. Humans depend on microbial cobalamin, usually obtained second-hand through consumed animal-derived products, for their cobalamin requirement. Multicellular plants or fungi do not synthesize or accumulate cobalamin and thus do not contribute it to the diet. Many prokaryotes synthesize and utilize corrinoids other than cobalamin, containing a lower axial ligand other than dimethylbenzimidazole or lacking a lower ligand entirely that cannot be utilized by humans. The human cobalamin transport system largely excludes these analogues from internalization by cells.
The estimated daily cobalamin loss is 1 µg in adults, with wide individual variations. Much of the loss is biliary. Estimates of daily intake requirement have varied, in part because of different end points for sufficiency. The Institute of Medicine reviewed available dietary data to set requirements for children of all ages, adults, and pregnant and lactating women ( Table 10-2 ). The recommended daily intake of cobalamin for adults is set at 2.4 µg/day, on the assumption that the expected 50% absorption rate of such an amount from food will replace the estimated daily losses while leaving a cushion for individual variability. A subsequent Scandinavian study of adults found that metabolic status leveled off with ascending daily intakes that approximated 4.2 µg or even 6 to 7 µg, but interpretations should be tempered by the limitations of the short-term dietary assessments for cobalamin, the biomarker end points, and the study populations (e.g., a striking overrepresentation of vegetarians in one study).
| Age Group | Recommended Daily Intake * (µg Cobalamin) | |
|---|---|---|
| Infancy | 0-6 mo | 0.4 |
| 7-12 mo | 0.5 | |
| Childhood | 1-3 yr | 0.9 |
| 4-8 yr | 1.2 | |
| 9-13 yr | 1.8 | |
| 14-18 yr | 2.4 | |
| Adulthood | 19-50 yr | 2.4 |
| Pregnancy | 2.6 | |
| Lactation | 2.8 |
* The amounts reflect recommended dietary allowances, which are derived from estimated average requirements by adding twice the latter’s coefficient of variation in order to include individuals whose requirements may lie at the extremes. For infants, however, the shown values represent adequate intake, based on evaluation of published data on breastmilk intakes that appeared to provide normal cobalamin status.
Analysis of NHANES data from 2003 through 2006 showed that median daily cobalamin intake was 6.4 µg (95% confidence interval [CI], 4.7 to 8.6 µg) in males and 5.0 µg (95% CI, 3.7 to 6.9 µg) in females. It is useful to note that the daily intakes reported by NHANES exceeded the Institute of Medicine’s recommendations in children (see Table 10-2 ). The intakes were 5.1 µg at ages 1 to 3 years, 6.1 µg at 4 to 8 years, 5.6 µg at 9 to 13 years, and 5.9 µg at 13 to 19 years. The 95% confidence intervals for these intakes exceeded the Institute’s estimated requirements, and the children’s 95% confidence levels for methylmalonic acid (MMA) levels were within the expected reference ranges. Data were unavailable for infants.
Foods vary widely in cobalamin content. Liver and some seafood are among the richest in cobalamin but are not eaten often, beef has relatively rich cobalamin content, and cobalamin content of eggs and milk is smaller. However, bioavailability can vary as much as tenfold among different food sources, and food cobalamin content, whose measurement can be imprecise, does not always predict effectiveness. Thus cow’s milk and fortified cereal, as well as fish, proved to be much more efficient providers of cobalamin than meat, despite the latter’s high cobalamin content. The specific receptor-mediated absorption of cobalamin via IF allows efficient absorption of more than 50% of this micronutrient while also excluding unutilizable corrinoid analogues. However, this mechanism is also easily saturable, and when more than 1 to 2 µg is ingested in a meal, much of the excess is relegated to nonspecific diffusion in which absorptive bioavailability falls to approximately 1%. The implications of such a small margin between the desirable level of intake and the total capacity of receptor-specific absorption have not been explored sufficiently. However, the existence of haptocorrin, a ubiquitous, relatively abundant, high-affinity cobalamin-binding protein that does not mediate specific delivery to intestinal cells or other tissue, raises the obvious question whether such an upper limit on cobalamin internalization serves a positive but still undefined biologic purpose, perhaps tied to corrin analogue exclusion from cells.
Cobalamin Requirement in Infancy.
Most of the information on cobalamin physiology and economy has been obtained in adults, which may apply to older children and adolescents also. However, data are limited in newborns and infants, who may differ considerably. Current estimates of the cobalamin requirement for infants are based on the 0.3 to 0.4 µg of cobalamin provided daily in breastmilk from mothers with apparently normal cobalamin status. However, recent data suggest that breastmilk may be a poor model. Cow’s milk contains not only much more cobalamin but its cobalamin is also carried by transcobalamin and may therefore be more bioavailable than the haptocorrin-bound cobalamin of human breastmilk.
Cobalamin Stores
Estimates of stores vary with assessment methods and subject selection. The current consensus is that cobalamin contents in adults average 2 to 3 mg, with wide spreads. The liver contains approximately 1.5 mg of those stores, with concentration ranges of 0.7 to 1.5 µg/g. By comparison, liver concentration is 0.02 to 0.14 µg/g in patients with megaloblastic anemia caused by pernicious anemia, which is a reduction of more than 80%. Store estimates are scarce in children. Maternal-fetal cobalamin transfer favors the fetus even when maternal stores are limited, and cord blood cobalamin levels exceed maternal serum levels, 370 versus 240 ng/L, although maternal serum cobalamin levels tend to be spuriously low during pregnancy. Full-term newborns have a mean liver cobalamin content of 27.3 µg ; the total liver content is only 10.7 µg in premature newborns and 4.1 µg in the fetus. The total body cobalamin store in newborns is approximately 50 µg, which illustrates how greatly stores need to expand between infancy and adulthood. As will be discussed, the approximately 1000 : 1 stores-to-daily intake ratio in adults and adolescents may be only 100 : 1 in newborns.
Physiology of Cobalamin
Cobalamin-Binding Proteins
Cobalamin does not exist free in the body outside the gastrointestinal tract, where its carrier-free existence is transient. Cobalamin traffic, transport, and access to cells depend on three cobalamin-binding proteins: IF, transcobalamin, and haptocorrin. Only IF and transcobalamin interact with specific cell receptors, allowing efficient cellular internalization of cobalamin at low concentrations, but these systems have relatively low capacity. These binding proteins have similar primary sequences, and their genes have similar intron/exon structure, suggesting that they arose by duplications of an ancestral gene. They show 33% overall homology with each other, and specific regions demonstrate as much as 60% to 80% identity.
IF is a 45-kD glycoprotein synthesized by gastric parietal cells in humans; under some circumstances, ectopic IF synthesis may also occur in chief cells, which are the normal synthetic site in many other mammals. IF mediates efficient uptake of cobalamin in the intestine. It binds cobalamins less tightly than does transcobalamin or haptocorrin but has far greater binding affinity for cobalamins than for nonfunctional corrin analogues, which helps limit the analogues’ intestinal absorption. IF is encoded by the GIF gene on chromosome 11q13.
Transcobalamin (also called transcobalamin II) is synthesized by endothelial and other cells, and mediates entry of cobalamin into most types of peripheral cells. It is a nonglycosylated protein with a molecular weight of 43 kD that, like IF, binds cobalamin tightly but has low affinity for cobalamin analogues. Transcobalamin content is small in many secretions. The TCN2 gene is on chromosome 22q12-13.
Haptocorrin (also called transcobalamin I) differs from IF and transcobalamin in having a high affinity for nonfunctional corrinoid analogues as well as for cobalamin and in having no specific cellular receptors for its internalization. These characteristics suggest an important role in excluding analogues, and perhaps excess cobalamin, from cells. The protein, encoded by the TCN1 gene on chromosome 11q12.1, is approximately 46 kD, but its heavy glycosylation creates glycoproteins of different sizes, isoelectric points, and plasma clearance rates. The glycosylation variability, which is sometimes secretion specific, led to artificial distinctions and names for haptocorrin variants (e.g., transcobalamin III and R binder), which are no longer used. Haptocorrin is synthesized in myeloid precursors at early stages when specific granules form ; this appears to be the chief source of plasma haptocorrin. Epithelial or ductal cells of exocrine glands also synthesize haptocorrin and are its source in secretions, where haptocorrin achieves concentrations much higher than in plasma. The secretions include bile, saliva, tears, breastmilk, bronchial secretions, amniotic fluid, and seminal fluid ; and their high haptocorrin content suggests a still unspecified role in relation to the gut microbiome and its abundant content of cobalamin and corrinoid analogues.
Typically, more than 70% of the cobalamin in plasma at any moment exists bound to haptocorrin, but this seeming preponderance is misleading because haptocorrin is cleared slowly from plasma ( ![]() , 9.5 to 12 days), whereas transcobalamin (
, 9.5 to 12 days), whereas transcobalamin ( ![]() , only 90 minutes) carries 40 times as much cobalamin daily in plasma as haptocorrin does. Study of haptocorrin interactions with each of the many corrinoid analogs has been difficult, but levels of plasma analogues, whose sources are unknown, approach or exceed cobalamin levels.
, only 90 minutes) carries 40 times as much cobalamin daily in plasma as haptocorrin does. Study of haptocorrin interactions with each of the many corrinoid analogs has been difficult, but levels of plasma analogues, whose sources are unknown, approach or exceed cobalamin levels.
Absorption of Cobalamin
Cobalamin in food is almost entirely protein bound. It is released by the action of acid and pepsin in the stomach, where it binds to salivary or gastric haptocorrin in preference to IF when gastric pH is low ( Fig. 10-4 , 1 ). In the duodenum, the haptocorrin-cobalamin complex from the stomach, and from bile, is broken down by pancreatic proteases, releasing cobalamin, which subsequently binds to IF ( Fig. 10-4 , 2 ). The IF-cobalamin complex binds to receptors in the apical brush border of enterocytes; the receptors are most abundant in the distal ileum ( Fig. 10-4 , 3 ). The absorbed fraction varies inversely with the amount of ingested cobalamin. Absorption efficiency is more than 50% when the amount is 1 to 2 µg, which approximates some meal contents. High intake in a meal or supplement (e.g., 5 to 10 µg) exceeds the capacity of IF receptors and the excess free cobalamin is internalized only by nonspecific diffusion, a process with only approximately 1% efficiency.
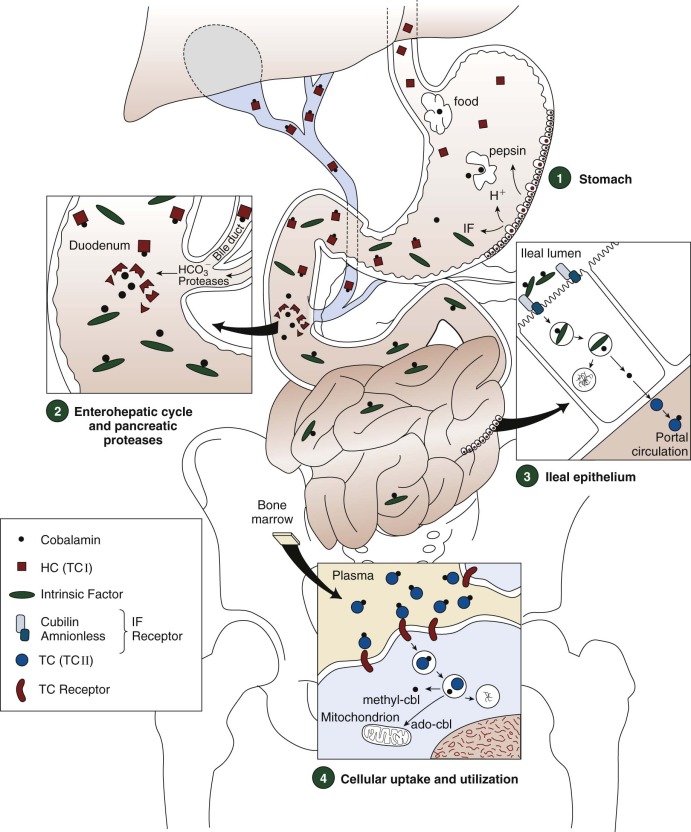
The ileal receptor consists of two proteins, cubilin (the product of the CUBN gene on chromosome 10p12.1) and amnionless (the product of the AMN gene on chromosome 14q32), and has been named “cubam.” Cubilin, a 460-kD protein that binds many ligands other than IF, such as transferrin and vitamin D–binding protein, is anchored externally to the enterocyte membrane by amnionless, a 48-kD membrane-spanning protein. Both components must be synthesized for cubam to be expressed at the cell surface.
The IF-cobalamin complex is rapidly internalized by endocytosis after binding to cubam. Cobalamin bound to transcobalamin (holotranscobalamin [holo-TC]) appears in the portal circulation 4 hours after ingestion. Although some studies have suggested that attachment of cobalamin to transcobalamin occurs within enterocytes after its release from IF, studies in transgenic mice were consistent with export of free cobalamin from enterocytes by the ABCC1 (MRP1) membrane transporter with subsequent attachment to transcobalamin outside the enterocyte.
Enterohepatic Recirculation
Approximately 1.5 µg of cobalamin and varying amounts of cobalamin analogues bound to haptocorrin are excreted in the bile daily. Cross-species experiments in rabbits suggest that plasma haptocorrin uptake by asialoglycoprotein receptors on hepatocytes delivers cobalamin and analogues to bile, but reservations exist and direct data are unavailable. Pancreatic proteases degrade the biliary haptocorrin (see Fig 10-4 , 2 ). Released cobalamin binds to IF and is reabsorbed in the ileum, whereas released analogues, unavailable to IF, are excreted in the feces. This process salvages about two thirds of the cobalamin secreted in bile.
Renal Cobalamin Reabsorption
The amount of cobalamin filtered daily is approximately 1.5 µg. Little cobalamin is detected in urine under normal physiologic conditions because more than 99.8% is reabsorbed in the proximal tubules. Cubilin and a second large multiligand receptor, megalin, are expressed in the proximal renal tubule, where they reabsorb a variety of filtered proteins. Filtered holo-TC is reabsorbed by megalin. Unbound cobalamin does not normally exist in plasma, but if it is introduced by parenteral injection, the filtered free cobalamin cannot be reabsorbed in the proximal tubule.
Cobalamin in Pregnancy
Cobalamin is efficiently transported across the placenta during pregnancy, and cobalamin concentration is higher in cord blood than in the maternal circulation, even in cases of maternal cobalamin insufficiency. The mechanism of cobalamin transport remains poorly understood. Both cubilin and amnionless are expressed in the visceral yolk sac of rodents, where they support uptake of nutrients (although not cobalamin). It is not clear that the visceral yolk sac plays any role in nutrition of the postimplantation embryo in humans. The transcobalamin receptor is present in human placenta, but its role in transplacental cobalamin uptake has not been studied.
Biochemistry of Cobalamin
Cellular Cobalamin Metabolism
Cobalamin functions as a coenzyme in two reactions in humans ( Fig. 10-5 ). MeCbl is required for methylation of homocysteine to form methionine, catalyzed by the cytoplasmic enzyme methionine synthase (EC 2.1.1.13). This reaction utilizes 5-methyltetrahydrofolate as methyl group donor and represents the point in cellular metabolism where cobalamin and folate metabolism intersect. AdoCbl is required in the conversion of methylmalonyl-coenzyme A (CoA) to succinyl-CoA, which is catalyzed by the mitochondrial enzyme methylmalonyl-CoA mutase (EC 5.4.99.2). In microorganisms, a variety of other enzymes utilize cobalamin for reactions that appear not to occur in mammalian cells.
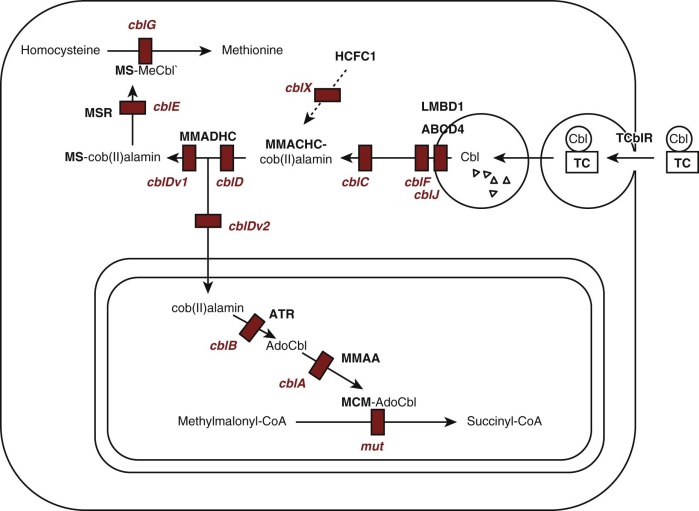
Intracellular cobalamin concentrations are low, and many of the forms that are involved in metabolism are not stable in aqueous solution. This has led to the suggestion that once cobalamin enters cellular metabolism it must be bound to protein at all times. Most cellular cobalamin exists bound to proteins, primarily methionine synthase in the cytoplasm and methylmalonyl-CoA mutase in the mitochondria. It appears that cobalamin is not retained in cells unless it is bound to protein.
Uptake of circulating holo-TC is mediated by the transcobalamin receptor, which initiates endocytosis (see Figs. 10-4 and 10-5 ). Transcobalamin is degraded in lysosomes, releasing cobalamin that is transferred across the lysosomal membrane by a process involving the lysosomal membrane proteins LMBD1 and ABCD4. Once in the cytoplasm, cobalamin becomes associated with the molecular chaperone protein MMACHC, which catalyzes reduction of the central cobalt to the Co 2+ state and removal of the upper axial ligand, if one is present. Subsequently cobalamin may enter the mitochondria, where cobalamin is converted to AdoCbl by cobalamin adenosyltransferase and becomes associated with methylmalonyl-CoA mutase; a third protein, MMAA, is required for transfer of AdoCbl from adenosyltransferase to methylmalonyl-CoA mutase. Alternatively it can remain in the cytoplasm, where it becomes associated with methionine synthase and is converted to MeCbl.
Biochemical Effects of Cobalamin Deficiency
The biochemical consequences of cobalamin deficiency result from decreased activity of the cobalamin-dependent enzymes methionine synthase and methylmalonyl-CoA mutase. Decreased activity of methionine synthase results in elevated levels of its substrate, homocysteine, in the blood and excretion of homocystine in the urine. Decreased activity of methylmalonyl-CoA mutase results in accumulation of MMA in the blood and urine.
Methionine Metabolism
Methionine is an essential amino acid in humans. Methionine synthase remethylates the homocysteine generated from methionine during cellular metabolism, helping to maintain adequate methionine levels as well as preventing accumulation of potentially toxic homocysteine. A cobalamin-independent enzyme, betaine : homocysteine methyltransferase, catalyzes homocysteine remethylation in liver, but its activity is insufficient to prevent homocysteine accumulation when cobalamin-dependent methionine synthase activity is impaired. Methionine levels, however, often remain normal in cobalamin deficiency.
Cellular methionine may be utilized in protein synthesis or adenosylated to form S-adenosylmethionine, which acts as a methyl group donor in several dozen different transmethylation reactions, including methylation of DNA, which plays a role in regulation of gene expression, as well as methylation of RNA, lipids, proteins (including histones), neurotransmitters, and thiols. These reactions generate the homocysteine that is acted on by methionine synthase. Homocysteine may also react with serine to form cystathionine, in a reaction catalyzed by cystathionine β-synthase.
Interaction of Folate and Cobalamin Metabolism
Deficiency of either folate or cobalamin, when substantial, produces megaloblastic anemia. The mechanisms leading to the anemia are complex. Folate deficiency directly affects the one-carbon substituted THF derivatives required for de novo thymidine and purine biosynthesis, resulting in abnormal DNA synthesis in bone marrow cells. The explanation for the hematologic similarity in cobalamin deficiency is believed to be the result of a secondary deficiency of these THF derivatives, best explained by the “methyl trap” hypothesis. Because methionine synthase is the only reaction in human cells that can convert 5-methylTHF to THF, which can be utilized to form one-carbon substituted THF derivatives, and because the reaction that converts 5,10-methyleneTHF to 5-methyTHF is irreversible under physiologic conditions, cobalamin deficiency results in “trapping” of folate in the form of now unusable 5-methylTHF.
Manifestations of Deficiency
Overview of Folate and Cobalamin Deficiencies
Cobalamin and folate deficiencies can cause similar clinical manifestations. The most frequent manifestation, megaloblastic anemia, is identical in both deficiencies and is therefore described here for both. Indeed, the megaloblastic anemia caused by cobalamin deficiency may respond initially if folic acid is mistakenly given, especially in doses above the bare minimum, although the hematologic improvement is often only partial and is rarely permanent. In contrast, the neurologic manifestations differ for the two vitamin deficiencies in adults, although cerebral and developmental consequences can be similar in infants with either deficiency. They will be discussed separately in sections devoted to each vitamin. Finally, the disorders causing each vitamin deficiency are very different. For all these reasons, rigorous differentiation between the two vitamin deficiencies must precede selection of the treatment.
Megaloblastic Anemia of Folate and Cobalamin Deficiencies
Three key features distinguish megaloblastic anemia from other anemias. First, there are the distinctive morphologic changes. Cells are not only macrocytic but their nuclei are also often larger. More specifically, a maturational nuclear-cytoplasmic dissociation defines megaloblastosis: the retarded chromatin condensation imparts an unusually immature, dispersed appearance to the nucleus, whereas the cytoplasm appears unremarkable ( Fig. 10-6 ). Myeloid cells have megaloblastic nuclei that can be abnormally shaped and too large for their already large cells (“giant” metamyelocytes and band cells). Megaloblastic changes in mature neutrophils are characterized by nuclear hypersegmentation, which is usually defined by the presence of at least one six-lobed nucleus or suspected when several five-lobed neutrophils are seen in a blood smear. Lobe-count averages above 3.0 to 3.3 lobes per neutrophil also suggest megaloblastosis, but lobe counts are tedious and their subjectivity requires that each observer establishes a personal normal range. When florid, megaloblastic changes are dramatic and obvious in peripheral blood and marrow (see Fig. 10-6 ) but mild changes in the early stages of deficiency are easily missed.
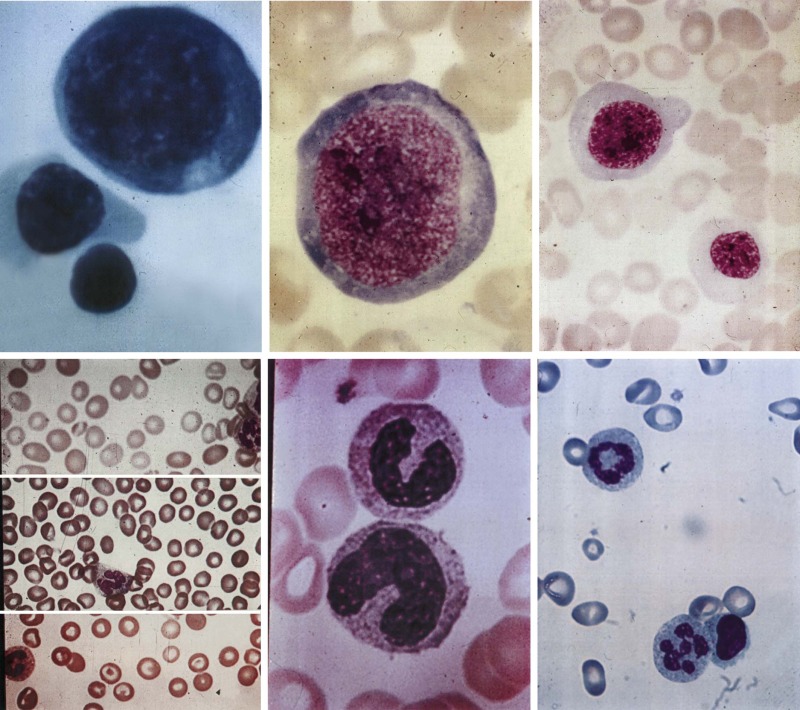
Second, megaloblastic anemia is the prototype of ineffective hematopoiesis. Precursors are actively generated in the bone marrow, but they are defective. As the dyspoiesis worsens, precursors increasingly undergo cell death in the bone marrow and are phagocytosed before they can exit into the blood as reticulocytes. As erythropoietin secretion increases, the bone marrow becomes more hypercellular, but the reticulocyte count fails to increase. Evidence of the intense intramedullary cell death includes increases in markers of cell turnover, such as serum lactate dehydrogenase, and the consequences of increased hemoglobin breakdown, such as indirect hyperbilirubinemia and decreased or absent haptoglobin. The decreased survival of megaloblastic RBCs in the blood adds an element of peripheral hemolysis.
Finally, the megaloblastic process affects platelets and myeloid cells also. The anemia is the earliest and, for some time, the only cytopenia. However, as the megaloblastic anemia progresses, granulocyte and platelet counts decline, and pancytopenia can eventually mimic aplastic anemia. In rare cases, an erythroblastopenia mimicking pure red cell aplasia may be superimposed.
Incidence and Evolution of Megaloblastic Anemia
Although the megaloblastic anemia is identical in folate and cobalamin deficiencies, its appearance after the dietary or malabsorptive onset of vitamin depletion in adults is measured in 2 to 3 months for folate deficiency but in years for cobalamin deficiency because of their disparate stores-to-daily intake ratios. The time course has not been documented as well in children, and onset may occur sooner in infants because of their smaller vitamin stores.
The earliest RBC changes are a rise in mean corpuscular hemoglobin followed shortly by a rise in mean corpuscular volume (MCV). These cell content and size changes precede the overt evidence of anemia, in part because the early phases of the anemia involve declining RBC count but little change in blood hemoglobin and packed cell volume levels, which are compensated by the increasing RBC sizes and contents. Clinicians must pay attention to changes in RBC indices regardless of hemoglobin and packed cell values. Although macrocytosis has many causes ( Table 10-3 ), early megaloblastic anemia must be considered whenever a patient’s MCV, even if still within the normal reference interval, has changed as little as 3 to 5 fL compared with a previous value. The onset of macrocytosis can be documented even sooner by reticulocyte-specific indices of macrocytosis. However, macrocytosis is sometimes masked if a microcytic disorder, most often iron deficiency or thalassemia trait, coexists. These combinations can produce normal, low, or high MCV, but the MCV is always higher than normal for that particular patient and it always decreases after the megaloblastic anemia is treated. The frequency of nonelevated MCV was 7% in one study of adults with pernicious anemia. The macrocytosis typically features macroovalocytes, which suggest but are not specific to megaloblastic anemia. Anisocytosis and admixture of macrocytes with normocytes elevate the relative distribution of RBC width. As megaloblastic anemia progresses, poikilocytosis, including teardrop cells, becomes prominent. With severe anemia, circulating nucleated erythroid precursors, Cabot rings, and Howell Jolly bodies can appear.
| Causative Conditions | Accompanying Hematologic Features |
|---|---|
| Megaloblastic Anemia | |
| Cobalamin deficiency Folate deficiency Antifol drugs (e.g., methotrexate) Cytotoxic drugs (e.g., hydroxyurea; 5-FU) Immunosuppressive drugs (e.g., azathioprine) Thiamine-responsive anemia Hereditary orotic aciduria | Megaloblastic changes, including hypersegmented neutrophils Macrocytosis can become severe Mild reticulocytopenia Pancytopenia (when the megaloblastic process is severe) |
| Disorders of Erythroid Production | |
| Aplastic anemia; PRCA; Blackfan-Diamond anemia Some sideroblastic anemias CDA; non-CDA dyserythropoiesis; Fanconi anemia Myelodysplasia; myeloproliferative diseases | Nonmegaloblastic Some disorders feature dyserythropoiesis and sometimes hyposegmented neutrophils Macrocytosis can often be severe (e.g., aplastic processes) Reticulocytopenia (often severe) |
| Reticulocytosis | |
| Chronic hemolytic anemia | Nonmegaloblastic; no hypersegmented neutrophils |
| Drugs and Toxins | |
| Alcohol abuse Some antiviral drugs (e.g., nucleoside RT inhibitors) Some anticonvulsant drugs | Mechanism of macrocytosis is often unknown No hypersegmented neutrophils |
| Nonhematologic Diseases | |
| Chronic liver diseases Hypothyroidism Copper deficiency | Nonmegaloblastic; no hypersegmented neutrophils Macrocytes are rarely oval |
| Artifacts | |
| RBC clumping by cold agglutinins; some warm RBC antibodies Severe hyperglycemia Hyponatremia | Nonmegaloblastic; no hypersegmented neutrophils Disparity between high MCV and normal morphologic examination |
The other early manifestation preceding anemia is nuclear hypersegmentation in neutrophils (see Fig. 10-6 ). It is also a much more specific feature of megaloblastic change than is macrocytosis. Unexplained hypersegmentation and other dysplastic granulocytic changes may also follow high-dose corticosteroid treatment of idiopathic thrombocytopenic purpura, however. The mechanism of hypersegmentation is unknown.
Neutropenia and thrombocytopenia are late features, appearing only infrequently before the hemoglobin level decreases below 10 g/dL; cytopenias that accompany mild anemia usually have independent explanations. The pancytopenia can ultimately become severe enough to mimic severe aplastic anemia. The two can be differentiated by the serum lactate dehydrogenase elevation (often exceeding 1000 units) and indirect hyperbilirubinemia in megaloblastic anemia.
Macrocytic anemia, the most common clinical feature of cobalamin or folate deficiency in adults, can be mild but it affects more than 70% of patients with clinically expressed deficiency, such as in pernicious anemia ; comparable data are not available in children. The reason why some patients with pernicious anemia do not develop anemia, even if they may have mild megaloblastic changes in the bone marrow that often escape detection, is unknown. Interestingly, a roughly inverse relationship between the severity of the hematologic and neurologic expressions has been described and the dominant expression tends to dominate again with relapses. A comparison of predominantly hematologically versus neurologically affected patients showed the predominantly anemic patients to have lower plasma glutathione, S-adenosylmethionine, cysteine, and folate levels, whereas a higher cysteine level was the most significant predictor of neurologic dysfunction.
Biochemistry of Megaloblastic Anemia
In deficiency of either folate or cobalamin, all dividing cells, especially bone marrow precursors and epithelial cells, show megaloblastic changes. MethyleneTHF, which mediates the methylation of deoxyuridylate to deoxythymidylate (see Fig. 10-2 ), is directly depleted in folate deficiency but can also be secondarily depleted in cobalamin deficiency, as best explained by the methylTHF trap hypothesis. Nevertheless, de novo thymidylate synthesis disruption, although necessary, may not by itself suffice to produce megaloblastic changes.
The misincorporation of excess uracil into DNA may exceed the capacity of excision repair, leading to single-strand breaks as well as occasional double-strand breaks. The latter may help explain the nuclear defects. Arrest at various stages of interphase occurs in megaloblastic precursors even while maturation proceeds, but the details are still unclear.
Mistaken folic acid therapy for clinically expressed cobalamin deficiency can bypass the methylTHF trap and improve the megaloblastic anemia. However, the response is often only partial and is usually impermanent. Concerns exist that neurologic manifestations may progress in the meantime and become irreversible as the cobalamin deficiency remains unrecognized and untreated. Systematic evidence is unavailable, and mistaken folic acid therapy may occasionally even improve neurologic status. However, ethical constraints prevent prospective studies and it is agreed that folate treatment must be avoided in cobalamin deficiency. These concerns prompted restriction of folic acid doses, as well as folic acid–fortified food intake, to 1 mg/day or less.
Ancillary Features of Megaloblastic Anemia
The ineffective hematopoiesis of megaloblastic anemia disrupts iron metabolism, raising serum iron, transferrin receptor, and ferritin levels, along with sideroblast counts and the ferritin content in erythroid precursors and macrophages. These changes can occur even in the presence of a coexisting iron deficiency, which may remain hidden until the ineffective erythropoiesis is corrected. Severe megaloblastic anemia also has a still unexplained element of intravascular hemolysis. Membrane protein changes, including spectrin, have been described in megaloblastic RBCs, but their impact is unknown.
Nonhematologic Manifestations of Cobalamin Deficiency
Neurologic Manifestations
Neurologic dysfunction is common, but its frequency is unclear because neurologic disease is not easily quantified, unlike the probably more common hematologic abnormalities. Whereas 13% to 27% of adults with pernicious anemia have had neurologic symptoms without anemia, the proportion with anemia without neurologic abnormalities is higher.
The classic neurologic manifestations feature a symmetric myelopathy, largely limited to the posterior and lateral columns (“subacute combined degeneration”) and neuropathy. The myelopathy typically begins with sensory and proprioceptive changes in the feet and ascends at variable rates to involve hands and arms as deficiency progresses. If not caught early, the changes may become partially or completely irreversible and can evolve to spasticity, ataxia, and incontinence. Other manifestations include mental changes of varying severity (mild electroencephalographic abnormalities can occur even without overt symptoms), autonomic dysfunction, and, rarely, optic neuritis.
In newborns and infants, deficiency and its manifestations appear sooner than in adults because of the smaller storage reserves, because hereditary defects of cellular metabolism often act more quickly and directly on tissues than dietary or malabsorptive disorders, and most especially because the developing nervous system is at greater risk than the mature nervous system. Often, the manifestations include impaired growth and development. Thus the neurologic changes common to older children and adults are overshadowed in infants by failure to thrive, poor brain growth, developmental delay, seizures, motor retardation, and movement disorders ; the movement disorders, which sometimes increase transiently after cobalamin therapy, can be mistaken for seizures. The diagnostic connection to cobalamin deficiency is often diverted or delayed for months and, even in nonhereditary disorders, developmental and neurologic residua may persist long after therapy.
The mechanisms responsible for the demyelination in the central nervous system, followed by axonal degeneration and gliosis, in clinical cobalamin deficiency, or for the nondemyelinative peripheral neuropathy, are unknown. Still unproven etiologic hypotheses for neurologic dysfunction have included abnormal myelin lipid synthesis because of abnormal propionate metabolism, defective neurotransmitter metabolism, corrinoid analogue accumulation, and cytokine abnormalities. Evidence from the clinical patterns in diverse cbl mutations and other findings favors a central, but perhaps not sufficient, role for methionine synthase inactivation, which disrupts methionine metabolism and S-adenosylmethionine–dependent methylation reactions. However, folate deficiency, which also inactivates methionine synthase, rarely produces neurologic abnormalities of comparable severity to cobalamin deficiency in adults, although hereditary disorders of folate metabolism in children often do.
Other Clinical Manifestations
Cobalamin deficiency can cause atrophic glossitis and stomatitis, which can be painful. Somewhat similar changes occur in folate deficiency, but the oral changes respond only to the specific vitamin whose deficiency caused them. Unexplained weight loss, sometimes substantial, can occur with severe cobalamin deficiency in adults and children, and it reverses after cobalamin therapy. Infants sometimes feature prominent gastrointestinal dysfunction, such as diarrhea. Severely deficient infants can also present with recurrent infections that mimic immune deficiency states.
Young patients with severe deficiency, especially nonwhites, sometimes display pigmentary changes that reverse with cobalamin therapy: skin hyperpigmentation that overlies joints, dark hair that turns a light reddish hue, or blue discoloration of fingernails and toenails. The pigmentary changes should not be confused with the unrelated, irreversible autoimmune vitiligo that occasionally coexists with pernicious anemia. Adults with pernicious anemia display reduced levels of osteoblastic proteins, such as osteocalcin and bone alkaline phosphatase, which respond to cobalamin. Reports of clinical bone complications in adults await systematic proof. Bone effects have not been studied in children.
Subclinical Cobalamin Deficiency
Unexplained low serum cobalamin levels that were unaccompanied by clinical manifestations, did not progress to clinical manifestations, and displayed no clinical benefits after cobalamin treatment were originally regarded as spurious. However, biochemical studies established that some of these represented mild metabolic cobalamin insufficiency and, unlike more than 90% of cases with megaloblastic anemia and other clinical dysfunction, they were rarely associated with the loss of IF or IF-mediated absorption by ileal cells. This often purely biochemical abnormality became known as “subclinical cobalamin deficiency.” As accurate, sensitive, automated tests of cobalamin metabolism, such as MMA and tHcy, became widely available, epidemiologic studies established that subclinical deficiency affected many adults, especially the elderly, and indeed was several times as common as pernicious anemia and other clinical deficiency states. The causes of subclinical deficiency are often unknown, but a plurality of 30% to 50% of patients have malabsorption of food-bound cobalamin, which is usually associated with chronic gastritis, achlorhydria, or gastric surgery. Because poor dietary intake alone cannot usually deplete cobalamin stores sufficiently in adults and older children to cause clinical deficiency, most vegetarians, especially adults, display only subclinical deficiency.
The health consequences of subclinical cobalamin deficiency are unknown, and progression to clinically expressed deficiency seems infrequent. In the absence of compelling data from randomized clinical trials, the benefits of intervention to reverse isolated biochemical changes of still unproven likelihood of progression to clinical deficiency remain unsettled.
Subclinical deficiency has not been studied in children and was believed rare. However, a transient, purely metabolic cobalamin deficiency affecting many normal infants in the first year of life has been documented in Scandinavia and is discussed in the section on dietary cobalamin insufficiency. The metabolic phenomenon must also be borne in mind when using current cobalamin status–related reference intervals in infants, and hematologists and geneticists must consider this transient phenomenon when evaluating MMA elevations in infants.
Nonhematologic Manifestations of Folate Deficiency
Neurologic Manifestations
Folate deficiency in adults does not cause the severe demyelinative myelopathy characteristic of cobalamin deficiency. Forgetfulness and mood changes, such as irritability, have been described, as has neuropathy. However, their attribution and specificity remain unsettled because folate deficiency is often not an isolated deficiency. Nevertheless, the neurologic consequences can be severe in some folate deficiency disorders in newborns and infants. Children with hereditary folate malabsorption, cerebral folate deficiency, and severe MTHFR deficiency (the last two of which do not display megaloblastic anemia) often have seizures, microcephaly, developmental delay, mental retardation, and myelopathy.
Other Clinical Manifestations
Mucosal changes of the tongue and mouth can sometimes resemble those of cobalamin and other deficiencies. However, because folate deficiency is often part of a broader nutritional inadequacy, responsiveness specifically to folic acid provides the best proof of causation.
Subclinical Folate Deficiency
Subclinical deficiency, definable as isolated abnormalities of folate and tHcy levels without megaloblastic changes or other clinical expressions of folate deficiency, has not been studied or defined as formally as subclinical cobalamin deficiency. Certain subpopulations are at special risk because of marginal dietary folate intake, increased folate requirements, or both, such as children (including adolescents), pregnant women, people with homozygosity for common polymorphisms of the MTHFR gene, and alcoholics. Although not studied formally, subclinical folate deficiency is almost certainly more common than clinically expressed deficiency.
Countries such as the United States and Canada have mandated fortification of cereal and grain products with folic acid to reduce NTD risks. This policy has nearly eliminated clinical folate deficiency in the general populations, sharply reduced the prevalence of isolated low folate levels, and lowered tHcy levels. The influence of folate status on hyperhomocysteinemia and on NTDs is clear, but the record on vascular disease, cancer, and cognitive decline in the elderly is mixed or ambiguous, as discussed elsewhere in the chapter. It merits emphasis that many of the public health risks, including NTDs, have generally been associated with subclinical folate deficiency and possible genetic or acquired alterations of folate demand or utilization, rather than with clinically overt maternal folate deficiency.
Diagnosis of Deficiency
Once detected, megaloblastic anemia and other clinical expressions of deficiency require confirmation that a vitamin deficiency is responsible and differentiation between folate and cobalamin deficiency. Measuring vitamin levels directly, the usual way to accomplish this, must be done before instituting therapy. Measuring metabolic biomarkers of vitamin adequacy, such as MMA and tHcy, is valuable whenever vitamin levels or the clinical picture are equivocal. Both metabolic biomarkers must be tested whenever a hereditary metabolic disorder that may not depress serum vitamin levels is suspected.
Biochemical tests are now accurate, automated, widely available, and generally quite sensitive, especially when the deficiency is advanced enough to have produced macrocytic anemia or neurologic symptoms. However, none of the tests qualifies as a diagnostic “gold standard,” largely because all of them lack the requisite diagnostic specificity for either folate or cobalamin deficiency. The innate specificity shortcomings have been exacerbated by inconsistent and sometimes controversial selections of cutpoints that demarcate “normal” from “abnormal” results, choices that have major, but often hidden, impact. Clinical judgment must always take precedence whenever a test result conflicts with the clinical picture. Pediatricians must also be aware that ranges of biochemical biomarker values appear to fluctuate during childhood, especially in early infancy and perhaps adolescence, where the fluctuations may sometimes hide subclinical deficiency rather than reflecting normal physiologic variability ( Table 10-4 ).
| Age | Cobalamin (ng/L) | Folate (µg/L) | tHcy (µmol/L) | MMA (nmol/L) |
|---|---|---|---|---|
| 4 days | 425 (322-634) | 11.9 (9.0-16.0) | 6.2 (5.0-7.5) | 290 (240-390) |
| 6 wk to 6 mo | 294 (199-393) * | 13.9 (9.4-19.1) * | 7.5 (6.1-9.2) * | 780 (360-1510) * |
| 1 to 10 yr | 747 (618-925) | 6.6 (5.3-9.3) | 5.2 (4.7-6.0) | 130 (110-170) |
| 10.5 to 15 yr | 591 (400-717) | 5.3 (4.0-6.7) | 6.5 (5.7-7.8) | 170 (130-220) |
| 15.5 to 19 yr | 500 (398-612) | 4.3 (3.4-5.6) | 7.8 (6.6-9.1) | 140 (120-180) |
* These values reflect distortions by the high frequency of transient subclinical cobalamin deficiency in breastfed infants. Breastmilk, although relatively rich in folate, can be limited in cobalamin content. (Cow’s milk is rich in cobalamin.)
The biochemical tests can also help evaluate the response to therapy. Measuring MMA or tHcy, depending on the deficiency in question, 1 to 2 weeks after therapy begins is useful because these metabolic intermediates improve only if therapy was effective. In contrast, serum vitamin content (i.e., cobalamin, holo-TC, and folate levels) rises automatically with vitamin therapy irrespective of therapeutic effectiveness. The timing of blood sampling relative to the most recent dose also determines the degree of automatic vitamin content change.
Vitamin Content Assays
Cobalamin Assay
Serum cobalamin measurement, although problematic, is the traditional test of choice for diagnosing cobalamin deficiency. Microbiologic assay, based on growth of cobalamin-dependent microbes on exposure to the test serum, is unavailable outside select reference and research laboratories. Clinical laboratories, which adopted isotopic, competitive-binding assays in the 1970s, have come to rely almost exclusively on automated, competitive-binding, chemiluminescence-based immunoassays since the late 1990s. Most laboratories define subnormal levels as below 200 or 250 ng/L (150 or 200 pmol/L). However, some authors raised cutpoints to 300 to 350 ng/L to capture nearly all elderly patients with otherwise isolated, mild MMA and tHcy elevations. This redefined reference interval led to claims that 40% or more of the elderly have cobalamin deficiency, but most of the redefined cases lacked clinical or metabolic evidence of deficiency, in part because abnormal MMA and tHcy themselves lack specificity. The cutpoint expansion worsened the already poor diagnostic specificity of a low cobalamin level. Most laboratories continue to use the 150-ng/L cutpoint in children, but levels, as shown in Table 10-4 , fluctuate during childhood, especially in early infancy, when cobalamin stores are small relative to intake and when maternal influences dominate.
Adults have many causes of falsely low cobalamin levels, including levels less than 100 ng/L sometimes. A major cause is pregnancy, with levels decreasing below 150 ng/L in almost half of women by the last trimester. The explanation is uncertain but may reflect maternofetal cobalamin transfer in part. Metabolic or clinical evidence of deficiency is rare, and cobalamin treatment is unnecessary. The levels become normal spontaneously a few weeks after delivery.
Folate deficiency often decreases serum cobalamin levels also, which become subnormal in 10% of patients. Folate therapy restores cobalamin levels to normal, but alertness is warranted because occasional patients, especially those with gastrointestinal disease, have concurrent deficiencies of both vitamins. Interestingly, hepatic cobalamin content is also somewhat low in folate deficiency. Other conditions associated with unexplained low cobalamin levels include oral contraceptive use, obesity, infection with human immunodeficiency virus, and multiple myeloma. Falsely low cobalamin levels also accompany hereditary or, less often, acquired deficiency of haptocorrin. Finally, the likelihood of falsely subnormal cobalamin levels rises sharply when overly liberal cutpoints, such as 300 ng/L, are applied.
Elevated cobalamin levels are more common than low levels in adults and can occasionally hide a coexisting cobalamin deficiency. Examples relevant to children include defects in cellular cobalamin metabolism, in which serum cobalamin (and holo-TC) levels remain normal or increase; these metabolic defects can be hereditary or acquired, as with nitrous oxide toxicity. A troubling malfunction peculiar to modern chemiluminescence-based cobalamin assays often causes spuriously normal (or elevated) levels in IF antibody–positive sera from cobalamin-deficient patients with pernicious anemia. Children with the juvenile form of pernicious anemia are often antibody positive and thus probably susceptible to the assay error. Renal failure can also cause spuriously elevated serum cobalamin. Race affects cobalamin levels; blacks have significantly higher levels at all ages than whites or Asians. Homozygosity for a common polymorphism of the FUT2 gene, affecting fucosylation, also causes slightly higher cobalamin levels. Finally, clinicians faced with unexpectedly normal or high cobalamin levels must consider recent cobalamin therapy as a possible cause.
Red cell cobalamin is rarely measured today because levels depend largely on the proportion of reticulocytes, which are rich in cobalamin, rendering results unreliable.
Serum Holotranscobalamin Assay
Holo-TC, the cobalamin fraction attached to transcobalamin, contains 10% to 30% of the total cobalamin in plasma at any moment in time (although the very high turnover of transcobalamin obscures the fact that transcobalamin carries much more cobalamin daily than haptocorrin does). Because holo-TC is the only cobalamin fraction that is available for delivery to cells, it is regarded by some authors to be a more reliable indicator of cobalamin status than serum (total) cobalamin. However, head-to-head receiver operator characteristics-based comparisons have shown holo-TC to be only marginally superior to total cobalamin (areas under the curves of 0.66 to 0.90 vs. 0.60 to 0.86 in six studies) in recognizing deficiency defined by MMA levels ; its specificity and positive predictive value are as poor as that of serum total cobalamin, but the causes await careful study. A recent study reported low holo-TC levels in folate-deficient patients also ; as noted earlier in this chapter, serum total cobalamin and hepatic cobalamin levels are also low in folate deficiency. Holo-TC testing also suffers from considerable cutpoint variability.
It is unclear whether the 90-minute half-life of plasma holo-TC leaves its levels too susceptible to transient changes in cobalamin flow from the gut to reliably reflect cobalamin stores and status, which change slowly. Evidence of relatively decreased holo-TC levels in patients with pernicious anemia in clinical and metabolic remission suggests that daily absorption flux, not just cobalamin adequacy, influence holo-TC levels, whose turnover is so great. More studies are needed to identify relevant influences that explain the poor specificity of low holo-TC levels.
Serum Folate Assay
As with cobalamin, folate testing began with microbiologic assay and, after 25 years of isotopic competitive-binding assay, is currently done by automated chemiluminescent assay. Current cutpoints approximate 2.5 to 3.0 µg/L (5.7 to 6.8 nmol/L) in adults. Normal levels fluctuate during childhood, with higher levels in early infancy that decline thereafter, reaching low-normal values in adolescence (see Table 10-4 ).
Serum folate has high sensitivity for folate deficiency. However, specificity is poorer because the serum levels disproportionately reflect recently ingested and absorbed folate. Transiently poor intake lowers the serum levels within 1 to 3 weeks, long before actual deficiency of stores.
A relevant cause of serum folate elevation is clinically expressed cobalamin deficiency, such as pernicious anemia, which causes RBC methylTHF to increase; however, the poor retention of methylTHF in RBC rapidly produces low RBC folate, along with high serum folate levels in 20% of cases. It is unknown if mild or subclinical cobalamin deficiency affects folate levels similarly. Dietary fortification with folic acid in the United States raised serum folate levels, and the frequency of levels below 3 µg/L decreased from 18% to 0.2%. Plasma tHcy levels improved commensurately.
Red Blood Cell Folate Assay
RBC folate content does not change after the reticulocyte stage and is thus less susceptible to short-term fluctuation than serum folate. The RBC level is a 120-day reflection of folate status, not a transient 1- to 3-week snapshot. However, RBC folate is subnormal in 60% of patients with pernicious anemia because the methylTHF accumulation caused by the folate trap in cobalamin deficiency is poorly retained by the RBC. Measuring serum folate, which is low in folate deficiency but high in cobalamin deficiency, can resolve the confusion. Other distortions of RBC folate content include blood transfusion, iron deficiency (which elevates RBC folate), and hemolytic anemia (reticulocytes have increased folate content). The choice between RBC and serum folate testing has generated disagreement, and clinicians should base their choice on the diagnostic question being asked.
Cutpoint selection has varied considerably. In adults, RBC folate cutpoints defined by megaloblastic anemia were set at 140 or 150 µg/L but later cutpoints based on metabolic criteria approximated 190 to 200 µg/L. A study of normal Scandinavian children identified age-related variation, with highest RBC folate in newborns (breastmilk has high folate content) and a steady decline from the first 6 months to the lowest levels in adolescents.
Biomarkers of Metabolic Status
Methylmalonic Acid Assay
Current gas chromatography/high-performance liquid chromatography/mass spectrometry methods are accurate and, with automation, serum MMA assay has become increasingly available; urine assays are done less often. Serum MMA elevation has 98% sensitivity for clinically expressed cobalamin deficiency, although its sensitivity in subclinical deficiency is unknown because no diagnostic gold standard exists for comparison. Being the only test unaffected by folate deficiency, MMA assay is widely considered the best available metabolic test for cobalamin deficiency, but its specificity is limited ( Table 10-5 ). A survey in adults found that cobalamin status, renal status (MMA values correlate directly with serum creatinine, even within the normal range), age, and sex explained only 16% of the MMA level. Creatinine is the major known confounder and should be measured when MMA is elevated.
| Elevation of tHcy | Elevation of MMA |
|---|---|
| Acquired cobalamin deficiency * | Acquired cobalamin deficiency * |
| Hereditary cobalamin transport and absorption disorders * | Hereditary cobalamin transport and absorption disorders * |
| Nitrous oxide toxicity * | Nitrous oxide toxicity * |
| Probable subclinical cobalamin deficiency in first year of infancy * , † | Probable subclinical cobalamin deficiency in first year of infancy * |
| Some hereditary cbl mutations that impair the methionine synthase reaction ‡ | Some hereditary cbl mutations and MM-CoA mutase deficiencies ‡ |
| Renal insufficiency (borderline, mild, or severe) | Renal insufficiency (borderline, mild, or severe) |
| Acquired or hereditary folate deficiency; antifol drugs | — |
| Folate-related polymorphisms (e.g., homozygous MTHFR C677T polymorphism) § | — |
| Disorders of homocysteine metabolism unrelated to cobalamin or folate (e.g., cystathionine β-synthase) | — |
| Miscellaneous conditions: hypothyroidism, acute lymphocytic leukemia, psoriasis, “life style” factors | — |
| Vitamin B 6 deficiency ‖ | — |
| Drugs: anticonvulsants, trimethoprim, levodopa, cyclosporine, azauridine, niacin | — |
| Severe alcohol abuse | — |
| Assay of serum instead of plasma | — |
| Inappropriately low tHcy assay cutpoint for abnormality | Inappropriately low MMA assay cutpoint for abnormality |
| Unknown causes | Unknown causes |
* Some cobalamin-deficient patients may have only MMA or tHcy elevation, not both, for unknown reasons; this is seen especially often in mild subclinical cobalamin deficiency.
† The tHcy elevation in infancy presumably caused by exclusive breastfeeding is milder than the MMA elevation, and levels are often high normal rather than elevated.
‡ See sections on cobalamin mutations for the specific entities.
§ Folate deficiency and tHcy elevation may not become expressed in some cases unless dietary intake is also limited.
‖ tHcy levels are normal in most cases, but abnormality can be elicited by methionine loading.
Most laboratories use a cutpoint approximating 270 nmol/L (0.27 µmol/L) in adults, but cutpoint selection has varied between 221 to more than 400 nmol/L, with predictably inverse impact on the frequency of cobalamin “deficiency.” MMA reference intervals have been lower in children than in adults, but they fluctuate in early infancy (see Table 10-4 ). Many infants display transient MMA elevations, now believed to reflect a mild subclinical cobalamin deficiency among exclusively breastfed infants but which had distorted pediatric MMA reference intervals for years.
MMA is also an effective, albeit more expensive, biochemical tool for monitoring response in cobalamin deficiency. Levels begin to decrease several days after cobalamin therapy (but not after folate therapy) and become normal within 2 to 3 weeks.
Plasma Total Homocysteine Assay
Plasma tHcy elevation is highly sensitive for folate deficiency and nearly as sensitive as MMA elevation for cobalamin deficiency. The degree of tHcy elevation in severe cobalamin or folate deficiency can mimic that seen in severe cystathionine β-synthase deficiency. However, inability of tHcy to distinguish between folate and cobalamin deficiencies limits its diagnostic utility. The many influences that elevate tHcy include serum creatinine, age, gender, and, uniquely among tests of cobalamin and folate status, preanalytic influences, especially using serum instead of plasma and failing to separate plasma from cells quickly (see Table 10-5 ). Cutpoint selection also affects diagnostic reliability. Reference intervals for tHcy fluctuate during childhood, peaking during the transient subclinical cobalamin deficiency affecting many healthy infants (see Table 10-4 ). A second, mild tHcy elevation occurs during adolescence, presumably reflecting a downward trend in folate status.
The high sensitivity of tHcy assay for cobalamin and folate deficiency and its relative inexpensiveness provide two valuable diagnostic roles: a normal tHcy level largely rules out the presence of clinically relevant folate deficiency, and the tHcy assay is the most cost-effective metabolic monitor of response of either cobalamin or folate deficiency to specific therapy. tHcy begins to decrease within a week and becomes normal within 1 to 3 weeks.
Miscellaneous Tests
Plasma cysteine increases along with tHcy in cobalamin and folate deficiencies, but methionine levels show little change. Measuring 2-methylcitric acid provides little diagnostic advantage over MMA measurement. The deoxyuridine suppression test, an older, indirect means of evaluating thymidylate adequacy, allows diagnosis of folate and cobalamin deficiencies at the cellular level when specific vitamins are added to cells in vitro but requires bone marrow cells and is labor intensive. Formiminoglutamic acid excretion lacks specificity and is no longer used diagnostically.
Response to a Trial of Specific Therapy
Objective evaluation of response to either cobalamin or folic acid provides the ultimate diagnostic confirmation. It is especially helpful in patients whose diagnostic results conflict with clinical judgment or are inconclusive. A convincing therapeutic trial requires giving the suspected vitamin alone, preferably as a single dose, and giving enough vitamin to produce a response but not so much that therapeutic specificity is lost. Approximate suggestions in older children and adults include 10 to 50 µg of cobalamin or 100 µg of folic acid; parenteral injection is advisable if malabsorption is present or suspected. Trial recommendations are less uniform in young children and must be tailored by size, age, and the nature of suspected diseases.
If anemia was present, reticulocytosis should begin within 3 days and peak at 7 days. Potential sources of diagnostic error include using more than one hematinic or using large doses of folic acid, which may elicit a hematologic response even in patients with cobalamin deficiency. With the correct vitamin, tHcy or MMA levels begin to improve within a week and become fully normal within 2 to 3 weeks, except in some severe metabolic disorders ; neither metabolite improves if the wrong vitamin is given. Serum vitamin and holo-TC have little monitoring value because vitamin content almost always increases, even when therapy is ineffective.
Acquired Causes of Cobalamin Deficiency
The diagnostic task does not end with identifying the responsible vitamin deficiency. Many management and prognostic decisions require knowing the underlying disorder. This section discusses the acquired causes of cobalamin deficiency, generally following the sequence of events from intake to cellular metabolism (see Fig. 10-4 ). Hereditary disorders will be discussed in detail in separate sections.
An important distinction between folate and cobalamin physiologies helps explain a major difference between the two deficiencies’ frequencies and expressions. The two vitamins’ body stores–to–daily intake ratios are strikingly disparate, especially in older children and adults. This difference means that adult folate stores can become depleted sufficiently within 2 to 3 months to cause clinical deficiency whereas cobalamin store depletion in adults requires several years.
A comparison gleaned from Chanarin’s summaries in 1979 of the scarce data of stores in infants and the Institute of Medicine’s recent estimates of daily intake requirements provides a perspective on the reduced cushions for dietary adequacy in infants. The infants’ daily cobalamin intake requirements are one fifth of that for adults but stores are only one fiftieth as great in infants as in adults. For folate, the infants’ daily requirements are one fifth that for adults, whereas stores are one twelfth as great as in adults. These estimates suggest that infants’ stores, while reduced for folate, may be especially precarious for cobalamin.
The underlying causes of cobalamin deficiency are listed in Box 10-1 . In adults and older children, clinically expressed deficiency usually occurs only if the highly specific absorption system fails and if the failure is both substantial and persistent enough to nearly exhaust stores. Therefore severe malabsorption, the most common form being pernicious anemia (loss of IF), accounts for more than 90% of clinical cases of deficiency in adults and older children but not in newborns and infants.

Dietary Cobalamin Insufficiency
Dietary Insufficiency in Adults and Older Children
A narrowly focused cobalamin inadequacy, ranging from strict veganism to mild ovolactovegetarianism, in which dairy products and eggs are consumed, can produce cobalamin deficiency. However, the process of depleting 80% of the very high storage cushion found in adults and older children to induce clinical deficiency with megaloblastic anemia or myeloneuropathy takes years and requires uninterrupted dietary restriction. Indeed, depletion takes longer than in pernicious anemia and other malabsorptive diseases because vegetarians continue to reabsorb biliary cobalamin normally. Subclinical deficiency limited to mild abnormalities of serum cobalamin and MMA levels can be seen, but clinical deficiency is surprisingly infrequent, even in strict vegans, unless other events, such as malabsorption or increased metabolic requirements, supervene.
Dietary Insufficiency in Infants
Intake adequacy poses greater problems in infants than in older children. The much reduced stores–to–daily intake ratio in newborns requires expansion during infancy and the demands of growth and development. Despite maternofetal transfer of cobalamin that favors the fetus even when mothers have borderline cobalamin status, the small cobalamin stores at birth must then be expanded to accommodate the demands of growth, neurologic maturation, and cognitive development. In early infancy, much (and often, all) of the diet consists of breastmilk. The severity and duration of any maternal restrictions, whether in cobalamin transfer to the fetus, breastfeeding, or both, can influence the degree of deficiency in the infant. Indeed, maternal cobalamin status at delivery correlates significantly with the infant’s status not just at birth but also 6 months later. Breastmilk cobalamin content is also influenced by maternal status.
Mild Dietary Insufficiency in Breastfed Infants.
A series of Norwegian studies beginning in 2001 documented unexplained elevations of serum MMA levels in 60% of normal, asymptomatic 4- to 6-month-old Norwegian infants whose mothers were not cobalamin deficient. The MMA elevation exceeded 1000 nmol/L in 30% of the infants, and median serum MMA was 780 nmol/L in infants aged 6 weeks to 6 months, compared with 290 nmol/L in newborns and 130 nmol/L in children aged 1 to 10 years. As Table 10-4 shows, plasma tHcy was also higher in the 6-week to 6-month age group, although not as strikingly abnormal as MMA. Cobalamin levels tended to be low-normal or, in only 8% of cases, slightly subnormal. These biochemical changes, which could also be easily reversed with cobalamin supplements, usually normalized spontaneously after several months. The absence of clinical signs of deficiency suggests that these normal infants had subclinical deficiency.
The explanation for the transient cobalamin status changes in early infancy has now become clearer. A recent longitudinal Danish study documented an unexplained, apparently spontaneous decline in breastmilk cobalamin levels in healthy, omnivore mothers 4 months after giving birth, often despite their taking cobalamin supplements. The authors associated this decline in breastmilk cobalamin with the transiently worse cobalamin status in exclusively breastfed infants compared with infants eating mixed diets ( Table 10-6 ). At month 9, the breastmilk cobalamin content had improved spontaneously, as had the cobalamin status of the infants (whose diet had also become mixed). The mothers’ cobalamin status remained unchanged throughout these fluctuations of breastmilk cobalamin. It remains possible that additional factors play a role because breastfed infants who also ingest a mixed diet showed somewhat similar cobalamin findings as exclusively breastfed infants at 4 and 9 months. The findings are compelling, but their surprisingly high frequency in seemingly normal mothers and infants requires clarification. More studies outside Scandinavia are needed as well. It is also important to note that this transient cobalamin deficiency of early infancy is subclinical. Convincing clinical accompaniments, including changes in hemoglobin and MCV levels, have not been reported. Finally, it is noteworthy that plasma folate levels remained unchanged throughout these transient cobalamin deficiencies but decreased sharply after the age of 1 year and continued to decline throughout childhood (see Table 10-4 ). The relatively high folate content of human breastmilk provides breastfed babies significantly better folate status than nonbreastfed infants.
| Birth/2 wk Postpartum | At 4 mo | At 9 mo | |
|---|---|---|---|
| Mean breast hindmilk | 561 (760) | 214 (290) | 325 (440) |
| Cobalamin concentration (n = 25) | |||
| Paired mean plasma | |||
| Cobalamin levels: | |||
| Mothers (n = 60) | 295 (400) | 288 (390) | 310 (420) |
| Infants (n = 60) | 325 (440) | 177 (240) | 236 (320) |
| Mean serum MMA | |||
| Levels (nmol/L): | |||
| Mothers (n = 60) | 180 | 170 | 160 |
| Infants (n = 28) | 270 | 440 | 240 |
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


