Medullary thyroid cancer (MTC) is a well-differentiated neuroendocrine tumor arising from the calcitonin-producing parafollicular cells, also known as C cells. C cells are derived from neural crest cells that form the ultimobranchial bodies of the fourth and fifth branchial pouches. During thyrogenesis, the ultimobranchial bodies fuse with the thyroid laterally and as a result, C cells are concentrated in the upper poles of the thyroid. In addition to calcitonin, MTC may secrete carcinoembryonic antigen (CEA), adrenocorticotrophic hormone (ACTH), and occasionally somatostatin. Symptoms related to hormone hypersecretion such as diarrhea, flushing, or Cushing’s syndrome may be the presenting manifestation and are usually representative of advanced disease.
Medullary thyroid cancer accounts for 3% to 4% of all thyroid cancers, and up to 13% to 14% of thyroid cancer-related deaths. Unlike differentiated thyroid carcinoma (DTC) of follicular-cell origin, the incidence of MTC has been stable despite improvements in diagnostic and genetic tests.1 MTC can be hereditary and part of the multiple endocrine neoplasia type 2 (MEN2) or familial MTC (FMTC) syndrome in 20% to 25% of cases, but is more often sporadic (75% to 80%). Overall 10-year survival for MTC patients is 75%, and poor prognostic variables include older age (>65 years) and advanced American Joint Committee on Cancer (AJCC) stage at presentation.1,2
The diagnosis of sporadic MTC usually occurs during routine evaluation of a solitary thyroid nodule, and the incidence of MTC in nodular thyroid disease ranges from 0.3% to 1%. The mean age at diagnosis is 50 years, and although thyroid nodules are more frequent in women, sporadic MTC is diagnosed in both genders with equal frequency.1 There are no known risk factors for the development of MTC except for a family history of MEN2 or MTC.3
MEN2 was initially described by Dr Sipple in 1961 when he identified an association between thyroid cancer and pheochromocytoma (Pheo). Seven years later, Steiner et al4 recognized primary hyperparathyroidism (PHP) as an additional manifestation, and labelled the constellation of endocrinopathies as multiple endocrine neoplasia 2. MEN2 is inherited in an autosomal dominant pattern, and is composed of three clinical variants: MEN2A, MEN2B, and FMTC (Table 34-1).3,5 In addition to MTC, Pheo, and PHP, MEN2A is also characterized by Hirshsprung’s disease and cutaneous lichen amyloidosis. The clinical manifestations of MEN2B are different from MEN2A, and include MTC, Pheo, musculoskeletal abnormities such as marfanoid habitus, mucosal neuromas that affect the lips, tongue, and conjunctivae, and ganglioneuromas of the urinary and intestinal tracts. The diagnosis of FMTC requires MTC in at least four multigenerational family members without PHP or Pheo. In MEN2A/2B, the penetrance of MTC is nearly 95%, but may be lower in FMTC kindreds. All hereditary MTC variants are caused by germline mutations in the RET proto-oncogene.6
| FMTC | MEN2A | MEN2B | |
|---|---|---|---|
| Major manifestations | MTC | MTC, Pheo, HPT | MTC, Pheo, mucosal neuromas |
| Minor manifestations | Cutaneous lichen amyloidosis, Hirschsprung’s disease | marfanoid habitus, ganglioneuromas of urinary and intestinal tracts | |
| MTC risk classificationa | A | A–C | D |
| Most common RET codon mutations | 5, 8, 14, 15 | 10, 11, 13 | 14, 15, 16 |
Activating RET mutations are identified in up to 95% of MEN2A/2B patients, and about 88% of FMTC patients. The RET gene located on chromosome 10q is composed of 21 exons and encodes a transmembrane tyrosine kinase receptor that is expressed during development in cells derived from the neural crest, branchial arches, and urogenital tract.5 In hereditary MTC, the specific RET gene mutation correlates with both tumor behavior and likelihood of associated disease manifestations. For example, mutations at codon 634 are among the most commonly found in MEN2A patients, and are associated with a high risk of Pheo (up to 50%) as well as MTC that is categorized as intermediate-risk for aggressiveness (Table 34-1). Prophylactic thyroidectomy in patients with hereditary MTC reduces disease-related mortality, and its timing is directed by RET mutation risk classification.3,7–9 Because of this beneficial impact on disease outcomes, current guidelines recommend that RET gene mutation testing be offered to all patients with an MTC diagnosis.3,6,10
Mutually exclusive somatic mutations in RET, HRAS, or KRAS are identified in about 90% of sporadic MTC.11 RET M918T is the most common alteration (50% to 75%) and is associated with a high prevalence of lymph node metastasis. RET mutations in exons 10 (codons 611, 618), 11 (codons 630, 634), and 15 (codon 883) have also been identified.12,13 In sporadic MTC, RET mutations have been shown to correlate independently with persistent disease and decreased survival.14 In up to 20% of sporadic MTC, mutations within the common hotspots of HRAS (codons 61 and 117) and KRAS (codon 12) have been described (Table 34-2).15,16 Exome sequencing of 21,000 genes in sporadic MTC cases revealed additional isolated and nonreplicated somatic mutations of genes involved in spliceosome and DNA repair pathways.11
Although the pathogenesis of both MTC and DTC implicates RAS and RET mutations as initiating events, the microRNA (miRNA) expression patterns do not appear to overlap.17 The distinctive miRNA signature observed in MTC could thus potentially be used as a diagnostic adjunct for preoperative fine-needle aspiration biopsy (FNAB), particularly if miRNA dysregulation patterns can be correlated to disease biology and prognosis.12,18,19 Whether hereditary and sporadic MTC have different miRNA expression patterns remains controversial; however, if true, this would also help guide preoperative patient care.
Medullary thyroid cancer has multiple histologic variants. Differentiation may not necessarily impact biologic behavior, but may result in varying cytologic and histologic features that can overlap with other malignancies.15 Classic cytologic features of MTC include a dispersed cell pattern of polygonal, triangular, or spindled cells with characteristically eccentric nuclei, coarse granular (salt-and-pepper) chromatin, azurophilic cytoplasmic granules on May–Grünwald–Giemsa stain, and background amyloid (Table 34-2). Amyloid can be found in up to 80% of cytology specimens, is similar to thick colloid, and appears translucent to slightly pink on Papanicolauou stain. When present, amyloid is highly suggestive of MTC, but can also rarely be found in benign goiter. A microfollicular pattern may be present leading to an incorrect cytologic diagnosis of “follicular neoplasm.” Overall, FNAB can diagnose MTC with sensitivity 65% to 95% and a positive predictive value (PPV) of 85% to 100%.3,15 Therefore, FNAB can reliably indicate the need for thyroid surgery in histologic MTC, although MTC may not always be accurately diagnosed preoperatively.
The most common variants of MTC are the spindle-cell and oncocytic variants that share pathologic features with sarcoma or anaplastic thyroid cancer, and papillary thyroid cancer (PTC), respectively. On histologic evaluation, calcitonin is the most helpful immunohistochemical marker for MTC. Although false-positives can occur in oncocytic neoplasms, non-MTC neuroendocrine tumors, and thyroid paragangliomas, the likelihood of a misdiagnosis can be reduced when immunohistochemical evaluation for CEA, thyroglobulin, cytokeratin, and TTF-1 are also performed.15 C-cell hyperplasia is a precursor of MTC in patients with hereditary predisposition, but can also be nonneoplastic and found in association with chronic lymphocytic thyroiditis and Graves’ disease.20 When identified with seemingly sporadic MTC, multifocal disease and/or c-cell hyperplasia should lead to RET gene testing.
A simultaneous diagnosis of MTC and DTC can occur rarely, and coexistent DTC appears to be increasing in likelihood among MTC patients just as it is for the population overall.21 Treatment should be guided by each tumor’s staging and guidelines accordingly, and is frequently directed by the MTC lesion. Disease-specific mortality of patients with coexistent MTC/DTC does not seem to differ from patients with MTC alone.22
Most patients with sporadic disease are asymptomatic, although advanced local disease can present with a palpable neck mass and symptoms of tracheal or esophageal compression in up to 15% of patients.3 Clinically or radiographically apparent lymph node metastasis is present in up to 50% of patients at presentation, and distant metastasis is diagnosed in 5% to 10%. Distant metastasis most likely involves the liver, lung, and/or axial skeleton.
Flushing and diarrhea is most likely due to hypercalcitonemia but can also be a result of Pheo in patients with undiagnosed MEN2A. MTC can also cause clinical sequelae of cortisol hypersecretion due to ectopic secretion of ACTH or CRH in up to 5% of patients with Cushing’s syndrome, and is typically a hallmark of aggressive disease. Because of the debilitating effects of excess cortisol, which is the primary cause of morbidity in this subset of MTC patients, treatment is recommended even for patients with widely metastatic disease and includes multimodality therapy to decrease tumor burden in addition to medical therapy to reduce hypercortisolism.3
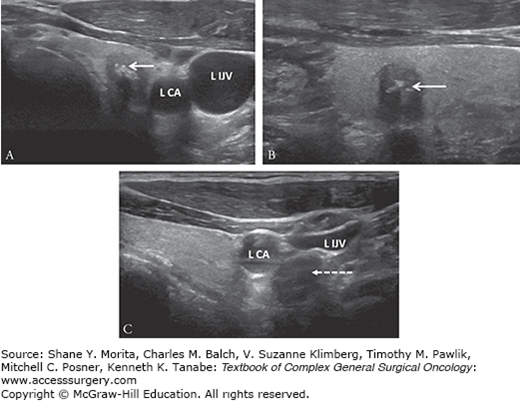
High-resolution neck ultrasound with lymph node mapping remains the gold standard imaging modality for thyroid nodule evaluation. Sonographic features associated with PTC have been well-described and include solid component, taller-than-wide shape, spiculated or ill-defined margin, marked hypoechogenicity, and intranodular micro/macrocalcifications.23 MTC has similar sonographic features except that MTC may be more likely to be ovoid-to-round in shape and to have macrocalcifications (Fig. 34-1).24,25 When MTC is suspected on ultrasound, FNAB is still standard management. Because of the high risk of lymph node metastasis at presentation, preoperative neck ultrasound must include assessment of bilateral cervical lymph node basins (levels II to VI) (Table 34-3).3,10 Rounded, enlarged lymph nodes without a fatty hilum should be biopsied for cytology assessment for documentation of positive cytology before recommending compartment-oriented neck dissection.
FIGURE 34-1:
Preoperative ultrasound images of medullary thyroid cancer (white arrow) demonstrating (A) transverse view of the left-sided 9-mm lesion that is hypoechoic and calcified, (B) with taller-than-wide shape that is more notable in sagittal view, and (C) associated with a sonographically suspicious 1.5-cm level IV lymph node (dashed line with arrow) located posterior to the left carotid artery (L CA) and left internal jugular vein (L IJV) that was nonpalpable. Metastatic multifocal medullary thyroid cancer was confirmed after total thyroidectomy, central compartment lymph node dissection, and left selective lymph node dissection.
| Imaging Test | Indication |
|---|---|
| Neck ultrasound | Preop: All patients for diagnostic evaluation of thyroid nodule, and lymph node mapping of central and lateral neck nodal basins Postop: All patients for surveillance |
| Neck CT | Preop: Clinical concern for locally advanced disease. N1 disease by neck US, or calcitonin >400 pg/mL Postop: Calcitonin ≥150 pg/mL Before reoperative surgery for curative intent |
| Chest CT | Preop: N1 disease by neck US, or calcitonin >400 pg/mL Postop: Calcitonin ≥150 pg/mL Before reoperative surgery for curative intent |
| MRI of liver or three-phase contrast-enhanced liver CT | Preop: N1 disease by neck US, or calcitonin >400 pg/mL Postop: Calcitonin ≥150 pg/mL Before reoperative surgery for curative intent |
| MRI of axial skeleton ± bone scan | Postop: Calcitonin ≥150 pg/mL |
| 18F-FDG PET or 18F-DOPA PET | Postop: Imaging negative, but elevated calcitonin/CEA |
Routine use of screening serum calcitonin levels as an adjunct to FNAB for thyroid nodule evaluation remains controversial (Table 34-4).3,10,26 Calcitonin screening of >10,000 patients evaluated during 7 years with nodular thyroid disease was reported by Elisei et al27; the prevalence of MTC was 0.4% and pentagastrin stimulation was used to confirm all elevations in basal serum calcitonin levels. Two patients had false-positive basal calcitonin levels but all remaining patients with elevated basal calcitonin levels had confirmed histologic MTC, and screening was more sensitive than FNAB.27 Furthermore, when compared to a historical cohort of MTC patients with longer follow-up, MTC patients who were diagnosed by calcitonin screening presented with an earlier stage of disease and had longer disease-specific survival. The added sensitivity of screening calcitonin levels to diagnose MTC preoperatively has also been demonstrated in other studies.28–30
| Pro | Con | Recommendations | |
|---|---|---|---|
| Screening | Increases sensitivity of FNAB for preop diagnosis of MTC MTC is diagnosed at earlier stage, and associated with longer survival | Cost-efficacy dependent on test cost and MTC prevalence Basal levels 20–100 pg/mL associated with high false-positive rate; pentagastrin or calcium stimulation improves PPV but thresholds are not well-defined | ATA/NCCN—no ETA CRN—yes |
| Staging | Preoperative basal serum levels correlate to disease volume, and direct extent of staging evaluation | — | Always check when MTC diagnosis known |
| Surveillance | Postoperative basal serum levels correlate to persistent disease Doubling time has prognostic significance | May take up to 6 months to reach lowest point CEA may be better marker for poorly differentiated MTC | Check 2–3 months after surgery, and draw serial levels to assess doubling time |
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


